

時間:

翻譯:華溶應用中心
審核:工業藥劑發燒友
體外藥物釋放試驗已成為半固體制劑開發和批準過程中最重要的工具之一。體外釋放試驗(IVRT)能夠反映多種理化特性、顆粒或液滴大小、黏度、物質微觀結構以及劑型聚集狀態的綜合作用情況。本文討論了IVRT的起源、原理及其與藥物動力學反應(如血管收縮或皮膚藥代動力學(離體皮膚))的等級關系,還討論了監管審批試驗要求的演變狀況。
IVRT能反映各種參數,是比較局部給藥制劑及以相似速率釋放相似量的活性成分的能力的逐級評價方式的重要部分。此外,除了能進行處方成分的定性和定量(Q1和Q2)考察外,IVRT還是評估局部給藥藥物分類系統(TCS)方法1類和3類藥物中提出的微結構排列(Q3)相似性的重要工具。本文討論了TCS系統以及局部給藥皮膚病藥物從Q1, Q2, Q3相同到Q1, Q2, Q3相似的發展概念,這個要求變化讓處方的調整變動有了更大的空間。
體外藥物釋放試驗目前已經成為半固體制劑開發和批準過程中最重要的工具之一。這是如何發生的呢?體外釋放試驗(IVRT)的起源是什么? 1987年,著名皮膚科醫生Richard Stoughton博士在《皮膚病學檔案》上發表了一篇文章,文章質疑了局部給藥糖皮質激素仿制藥與原研藥的等效性。這類出版物令監管機構感到不安,并且立即予以關注。從口服制劑評價方面獲得的經驗可知,溶出度在劑型的生物利用度中起著重要作用。固體口服制劑溶出度試驗在當時已經逐漸得到重視并建立了質控檢測,以確保產品性能和批間一致性。溶出度試驗的通用原則應適用于局部給藥制劑的體外釋放。與固體口服制劑的溶出類似,局部給藥劑型的藥物釋放對其質量、安全性和有效性也起著重要作用。對于局部給藥制劑,給藥后的第一步就是將有效成分從制劑中釋放出來,然后藥物分布、滲透或穿過角質層。根據活性成分的理化性質、處方因素以及預期作用部位的不同,特定的藥效作用可以在皮膚的特定的、最外層或更深層表達。對局部濃度的評估是復雜的,需要在適當的時間間隔下保持足夠的且持續的濃度。1989年在華盛頓舉行的“體內經皮滲透/吸收”會議上列清了每種可用的方法都有其優點和局限性。因此,研究人員決定探索局部給藥劑型的體外釋放方法開發。
許多在皮膚醫學領域工作的制藥行業和藥物研究人員都非常熟悉使用尸體皮膚的Franz擴散池(垂直擴散池,VDC)系統。該系統用于評估局部給藥藥物開發中有效成分的滲透和滲入情況。決定將該系統改良用于開發適應局部給藥半固體特性的體外釋放。尸體皮膚的使用受到許多變量的影響,不適合在質量控制檢驗程序中常規使用。因此,有必要開發一種利用VDC和具有更強重復性的市售合成膜的藥物釋放方法。
使用VDC和合成膜的體外藥物釋放方法被開發出來,并于1989年首次發表在《國際藥學雜志》上。局部用藥領域的研究在幾個中心進行了探索,包括美國食品藥品監督管理局(FDA)實驗室,以及法國的Hans Schaefer教授、密歇根大學的Gordon Flynn教授、羅格斯大學的Joel Jatz教授和猶他大學的Lynn Pershing博士,他們都對局部給藥制劑的IVRT和生物等效性(BE)的不同方面進行了評估。大部分研究結果隨后由所有研究人員發表。由于IVRT采用了一種新技術,FDA在德克薩斯州達拉斯市的實驗室為工業科學家們提供了幾次實踐培訓課程。
開發有意義的半固體體外釋放試驗的研究工作繼續進行,重點是開發一種簡單、可重復和靈敏的方法,在可行的情況下,具有一些已知的臨床相關性。在IVRT的初始開發過程中獲得的經驗表明,在保持方法簡單和可重復性的同時,很難模擬所有導致體內過程復雜性的因素。作為生理屏障,皮膚的厚度、結構、滲透性和對局部給藥處方的反應在個體內和個體間都是多變的。此外,在體內給藥后,劑型會發生形態變化、不同程度的組成和結構轉變,這取決于劑量、方法和給藥部位等因素。因此,很難開發一種能夠模擬臨床環境的標準化測試。與固體口服劑型相比,IVRT和局部給藥制劑建立體內外相關性被證明更具挑戰性。然而,兩種不同的氫化可的松乳膏的體外藥物釋放率(Caron et al, 1990)、兩種戊酸倍他米松乳膏產品的皮膚漂白和藥物釋放(Shah等,1992)、類固醇的效力和藥物釋放(Shah et al, 1999)與體內皮膚藥代動力學(DPK、膠帶剝離)和藥效學(血管收縮試驗、VCA或漂白效果)參數之間至少有一定的順序對應關系。很明顯,使用非皮膚的人工膜,惰性是關鍵要求,并且在封閉條件下使用高劑量(偽無限),而釋放的藥物在滿足漏槽條件的接收池中收集,這將無法預測生物利用度。在最初的工作中,重點是了解IVRT所反映的內容,比較已知成分和/或效力存在差異的產品的體外行為。體內數據對于證實一種簡單的方法至關重要,盡管臨床復雜性過于簡化,但能夠通過不相似的體外行為確認這些差異。IVR已經發展成為一種比較測試,它應該對局部作用產品的特性敏感,這對體內結果至關重要。這進一步意味著了解體外比較的產品(均質或異質體系,活性物質完全溶解或部分懸浮,其在體系組成相中的分布,釋放機制和限速步驟等),識別可能與臨床相關的差異,并應用適當的相似性標準。規格區分是首要要求之一。IVRT必須在給定制劑中顯示不同藥物濃度的不同釋放(Pillai et al, 2001)。IVRT受到了許多研究者、產業界和學術界的挑戰、反對和抵制。它并沒有被輕易接受。1997年,在SUPAC-SS指南中引入了IVRT,作為擴大規模和批準后變更產品一致性的衡量標準(FDA, 1997)。1998年舉行了FDA /美國制藥科學家協會研討會,討論IVRT的利弊。研討會總結了IVR方法的有用性(Flynn et al, 1999)。1998年第一個FDA局部給藥制劑指南草案強調了IVRT的重要性和價值(FDA, 1998)。因此,IVRT慢慢開始被接受。關于局部給藥制劑的指南草案后來因IVRT以外的原因被撤回(FR, 2002)。
體外釋放(IVR)方法的發展與體外溶出度試驗類似,應用于研究和開發階段,穩定性研究或變更或變異分析。它的作用是基于假定的能夠綜合反映諸如顆粒或液滴大小、藥物溶解度、流變性等幾個因素的影響。IVRT被納入FDA發布的非無菌半固體制劑規模擴大和批準后變更指南(FDA, 1997),用于評估已批準的成分或生產工藝的明確的二級(中等)變更。這一首次正式申請標志著一種謹慎的方法,在這種方法中,首先對兩種比較產品之間的變更進行定義、分析和分類,并根據它們在體內的潛在影響進行分類,然后通過能夠評估性能顯著變化的適當方法進行評估。上市的、變更前的產品作為參比,而自制的、變更后的制劑在組分或生產工藝都一定程度的不相似。1998年指南草案試圖擴大其在開發低規格局部給藥產品中的作用。對于新藥申請(NDA)的提交,指南草案指出,在“BA(生物利用度)文件”被認為重要的情況下,對于較低規格“可以執行IVR”。一旦仿制藥最高規格的生物等效性在體內得到證明,IVRT就有可能被用于免除較低規格的體內評估(FDA, 1998),前提是:i)兩種規格的組成的唯一區別是活性成分和相應稀釋劑的量;Ii)生產工藝和設備相同;Iii)參比制劑有相應的兩種規格上市。應用相同的方法獲得的IVR速率將在產品和規格之間進行比較。該指南草案于2002年被撤回(FR, 2002),原因是兩位研究者在DPK研究結果中觀察到不一致。然而,后來人們注意到,兩位研究者采用了不同的方案。指南草案中描述的IVRT相關問題沒有爭議,因為它的地位已經在SUPAC-SS中確立。
低規格的生物豁免標準現已包含在歐洲藥品監督管理局(EMA)于2018年發布的關于局部用藥質量和等效性的新指南草案中(EMA, 2018)。這說明了與體外溶出試驗的類似性,以及在局部半固體制劑的開發和評價中日益重要的作用。美國藥典(USP, 2013)第1724章首次描述了各種類型的擴散池。美國FDA發布了越來越多的產品個例指南(PSG)文件草案,其中包括體外選擇作為生物豁免的一種手段。被比較的兩種產品(試驗制劑和參比制劑)應具有相同的定性組成和相似的定量組成。根據劑型的復雜程度,IVRT是主要測試之一(1%磺胺嘧啶銀乳膏或5%阿昔洛韋軟膏的PSG;FDA, 2017年,2019年),或者在許多情況下,是更復雜的比較方法的一部分,該方法結合了幾種物理化學評估和通過人體皮膚樣品的體外滲透試驗(IVPT) (5%阿昔洛韋乳膏的PSG;FDA,2016)。只有IVRT有明確的接受標準,長期實施的SUPAC-SS指南驗證了這一點(FDA, 1997)。
體外釋放試驗當前被認為是保證產品質量的有價值的工具,并在PSG文件中找到了它的位置。體外釋放度反映原料藥和制劑的幾種理化性質的綜合作用,包括活性成分的溶解度和粒徑;這也是半固體制劑的組成和微觀結構特性綜合影響的一個很好的指標。生產方式和工藝可能改變制劑屬性,從而影響藥物釋放速率和藥物的生物利用度。當前版本的EMA局部給藥藥品質量和等效性指南草案(EMA, 2018)中指出,需要IVRT來支持擴展藥物等效性的概念。其他監管機構建議將其用于選擇進行體內測試的代表性批次藥品(NIHS, 2003)。
現已提供IVR方法開發和驗證的建議(FDA, 2016)。一般情況下,釋放介質的選擇是基于漏槽條件的要求,膜的選擇必須證明其惰性,人工膜作為被測半固體的機械支撐。介質和膜的組合應與制劑和活性成分相容,在整個試驗期間防止任何明顯的降解或結合現象。此外,它應該提供一個與半固體基質適當的接觸角,從而允許明顯釋放。具有已知優點和局限性的各種各樣的擴散池和現有的溶出裝置適應裝置是可用的。方法驗證包括規格-釋放速率關系的評估以及微觀結構差異的區分。
對于目前的所有應用,必須開發并使用IVRT作為比較評估,即比較速率,在某些情況下描述擴散過程的附加參數。生成的數據的相關性總是依賴于參比制劑的存在,需要對參比制劑進行適當的描述。兩種半固體藥品之間的相似性和差異性應根據其對質量、安全性和有效性的潛在影響進行識別和理解。比較表征的方案應從成分組成開始,然后應根據劑型的復雜性進行理化特性和微觀結構測試。值得注意的是,局部給藥半固體在整個保質期內不斷變化。隨著時間的推移,這種變化可能會給仿制藥的開發帶來額外的問題,參比制劑是一個移動的質量目標。IVRT由于其對一系列物理化學和微觀結構參數的敏感性,也應考慮這些動態變化。
IVRT最重要的應用是在局部皮膚科藥物審批過程中的地位。在某些情況下,它是仿制藥批準的基礎,在許多情況下,它是批準要求的一部分。目前,IVRT是多方面方法的一部分,經常結合根據劑型的特殊性和復雜性選擇的一系列比較的物理化學評估。當要比較多源產品時,對IVR數據的充分解釋取決于定性和定量組成的相似程度(由限制性標準定義)。根據EMA指南草案(EMA, 2018),這種由+/-5%差異定義的限制性(在某些情況下為+/-10%)可以通過對質量、安全性和有效性的潛在影響來解釋。進一步擴大IVRT作用的主要問題是,單獨的這項測試可能無法適當地反映超出這些限制的差異的臨床影響,特別是當注意到不同輔料會對體內結果產生影響時。IVR速率取決于測試條件,不反映滲透或滲入特性。然而,該速率從未被用作安全性和有效性的直接預測指標。IVRT一直是一種比較評價,它被認為反映了半固體基質的成分組成(輔料的種類、質量和用量)和成分排列的相似性和差異性的綜合效應。對這些差異的潛在后果進行深入分析,無法充分解釋IVRT結果。根據這一基本原理,提出了局部給藥藥物分類系統(TCS),如果被接受,將為許多仿制局部給藥產品提供生物豁免(Shah等人,2015)。
局部給藥藥物分類系統(TCS)是基于SUPAC-SS、成分定性(Q1)和定量(Q2)的一致性、非活性成分的作用、微觀結構排列(Q3)和IVRT對局部給藥仿制藥進行分類的框架。在TCS 1類仿制藥中,Q1和Q2與參比制劑(RLD)相同,Q3和IVRT與RLD相同,反映了SUPAC-SS的1級變更。這些仿制藥將有資格獲得生物豁免。在TCS 2類的情況下,仿制藥與RLD相比具有Q1和Q2,但不具有Q3,并且具有不同的IVRT特性。這可能是因為輔料來源和質量特性不同,也可能是由于生產過程中使用的方法和參數不同。在這些情況下,仿制藥不符合生物豁免資格,必須按照監管機構的要求進行BE研究。在TCS 3類中,當測試產品與RLD進行比較時,Q1和Q2是不同的,但具有功能相似的非活性成分,導致Q3和IVR與RLD相似。這些仿制藥將有資格獲得生物豁免。我們發現處方/產品流變學的變化總是反映IVR的變化,從而支持了確保變化下產品一致性的有效性和重要性。TCS 1類和3類仿制藥和生物豁免概念與公認的生物藥劑學分類系統(BCS)概念相當。在這兩種情況下,生物豁免是基于劑型的體外釋放或溶出結果提供的,并且對3類生物豁免的要求比1類更嚴格。針對TCS 3類產品,提出了一種基于風險的輔料評估方法。TCS分類不排除任何額外的物理化學測試要求。如果是TCS 4類,仿制藥的成分不同,導致微觀結構不同,IVR也不同,不符合生物豁免條件。簡而言之,根據TCS提案,1類和3類局部給藥仿制藥具有與RLD相似的釋放特性(通過應用SUPAC-SS定義的數據分析、比較程序和驗收標準)將有資格獲得生物豁免。2類和4類仿制藥將需要按照監管機構的要求進行體內BE研究。
仿制局部給藥皮膚科藥品現在被歸類為復雜仿制藥。這些產品的批準多年來一直在不斷發展,從要求仿制藥與RLD達到Q1、Q2并要求進行比較臨床終點研究(糖皮質激素除外),到Q3的一致性,與RLD相比,具有不同非活性成分的仿制藥只要不顯著影響藥物的局部或全身生物利用度就可以接受。在最近的監管報告中反映的2019年的這一變化是一個受歡迎的變化(Raney, 2019)。然而,應該指出的是,“相似性”的概念在2015年的TCS分類TCS 3類中被描述和提出(Shah et al, 2015)。TCS 3類中“類似Q3”的概念符合當前Q1、Q2、Q3相同向Q1、Q2、Q3相似的演變思維(Raney, 2019)。我們對已上市的5%阿昔洛韋乳膏產品的廣泛研究也支持了這一概念(Miron等,2021b)。
圖1顯示了TCS 3類和Q3相似概念的比較。局部皮膚科藥物概念的變化,即Q3相同演變為Q3相似,解釋為 Q1和Q2可能存在差異,但與參比制劑相比,它不應顯著影響局部或全身生物利用度。這顯然符合提出的TCS 3類的分類。
回顧SUPAC-SS指南成功應用超過25年,并作為當前局部給藥半固體性能測試應用的基礎,使用IVRT來評估TCS提出的Q3相似性或差異性是合理的。SUPAC-SS指南中描述的二級變更及其轉到潛在的Q1/Q2/Q3或TCS分類中表明,隨著成分和組成的變更或生產和工藝的變更,如果變更前和變更后的產品之間的IVRT相似,那么變更后的產品不懷疑對產品的體內性能有任何重大影響。表1總結了SUPAC-SS 2級成分組成變更與TCS 1、3類的關系。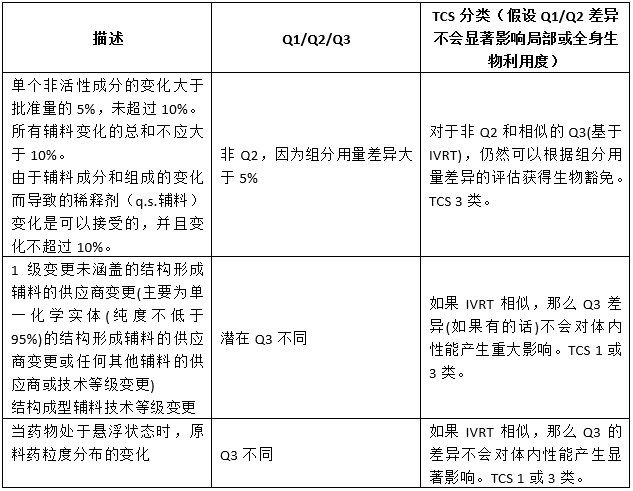
EMA基于比較仿制藥和參比藥的分步方法,闡述了擴展藥物等效性的新概念(EMA, 2018)。EMA表示,應該從深入了解局部給藥產品提供預期安全性和有效性的方式開始,制定具體的方案,而不是由US-FDA發布的PSG。它是從劑型的復雜性、給藥部位、相似的定性和定量組成的重要性、詳細的和產品特異性的比較理化測試、IVRT和IVPT,以及滲透動力學和藥效學甚至臨床治療等效研究(如適用)開始的逐步方法。沒有這些評估應說明理由。鼓勵采用新的方法。如前所述,“需要經過驗證的體外釋放試驗來支持擴展的藥物等效性”。
在當前版本的EMA指南草案(2018)中,與美國FDA相比,IVRT的一般原則相似。然而,在樣本量(每批產品進行比較評估的批次數、每批重復的次數)、試驗期結束時的劑量消耗、實驗數據與Higuchi模型擬合的標準、比較參數(累積釋放量、釋放率和滯后時間)、統計比較(包括IVR數據的假設分布)和相似性的接受標準等方面仍然存在顯著差異。
USP藥典第1724章的修訂版于2022年5月2日在藥典論壇上發表。在簡介部分,文件總結了一般測試原則以及將IVRT和IVPT作為產品性能測試的正確使用方法。列出了IVRT的局限性,以及當前的擴展應用,例如將“預期的仿制(測試)產品”與“可以在某些情況下支持生物等效性證明”的參比產品進行比較,或將“經過驗證的IVRT方法(…)作為將來產品參考的基礎”的體外釋放速率的潛在使用。典型的精度和可重復性對于性能測試來說是一個相當大的優勢,假設是“反映了處方中物質的物理化學性質和/或結構排列的差異”。IVRT的微觀結構敏感性在目標的表格摘要中提到,我們增加了預期的效用或相關性(表2)。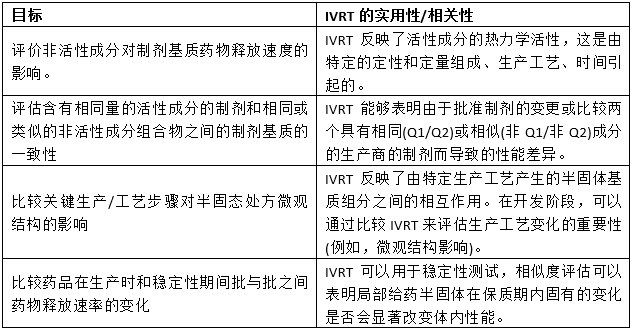
IVRT已經慢慢進入局部給藥半固體的開發和評價領域。當前版本的監管文件,無論是PSG還是指南草案,都證明了謹慎使用這種體外性能測試。它是作為一個復雜的、產品適合的、比較評估的一部分,在許多情況下,IVPT被認為是一個更相關的體內反映的方法。IVRT的更廣泛應用受到了批評。假設高度復雜的體內遞送和滲透過程的過度簡化和標準化不可能具有生物相關性。那么,IVRT是一種不反映體內性能的質量測試,還是一種只反映藥物質量的性能測試??SUPAC-SS指南從一開始就將IVRT與體內性能的潛在影響聯系起來。另一方面,將IVRT納入多源產品的比較評估表明,它不是一個多余的質量測試,而是一種評估,可以解決具有相似成分和微觀結構的產品之間生物不等效的額外風險。
在局部給藥藥品、生物等效性和IVRT的監管方面做出了重大努力。IVRT可能會成為半固體制劑的強制性規定,類似于口服固體制劑的溶出,以確保批間產品質量。IVRT作為一種產品開發工具和質量控制工具可以獲得更多的重要性,盡管在許多情況下,半固體基質在保質期內的內在變化是復雜和顯著的,這使得接受限度的設置特別具有挑戰性。最后,IVRT將成為生物豁免的工具:(i)低規格半固體制劑的批準和(ii)基于局部給藥藥物分類系統(TCS)的生物豁免,類似于BCS。
IVRT已經走過了漫長的道路,它在藥物開發和局部皮膚病藥物的評估中確立了自己的地位。IVRT作為一種生物豁免工具可能適用于評估劑型的低規格和TCS 1類和3類藥物。
略






