

時間:
摘要:闡述了通過科學地選取體外溶出度試驗條件,提高其與體內生物利用度的相關性,以及對藥物制劑工藝的要求。 結合我國目前固體藥物制劑發展現狀和存在的問題提出了建議。
關鍵詞:溶出度;生物利用度;生物等效性;藥物制劑;質量控制
中圖分類號:TQ460.7 + 2;R969 文獻標識碼:A 文章編號:1001-8255(2005)07-0447-05
對于藥物固體制劑,國產藥與進口藥有什么區別?為什么同樣劑型、同樣劑量的某些藥物,患者服用后會有不同的療效1?用什么試驗方法、什么檢測指標才能夠科學、有效地評價出國產藥與進口藥在臨床療效上的差別呢?難道一定要到最終臨床階段才能明了嗎?難道僅是因生產條件—— 即是否經GMP認證的生產條件所致嗎?
1 .體內環境對藥物吸收的影響
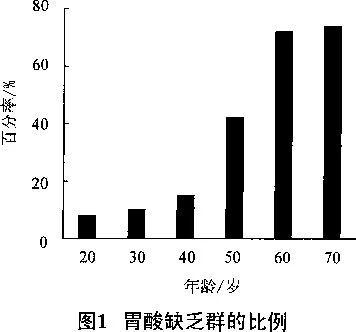
人體對藥物的吸收部位主要是消化道。體內環境正常者,胃腸道內存在有正常量和正常pH的胃酸和腸液;體內環境非正常或體質虛弱者,胃酸和腸液的量及pH 會有差異。據報道 2 ,人體消化器官內的液體pH 范圍為:胃 1.2~7.6,十二指腸 3.1 ~6.7,小腸 5.2~6.0。50 歲以上的人群,胃酸和腸液均有變化( 圖 1 ) 。
收稿日期:2005-05-08
作者簡介:謝沐風( 1973) ,男,碩士,從事新藥復核和質量標準的研究。2003.8~2004.2曾赴日本國立醫薬品食品衛生研究所薬品部進修。
Tel:021-64703139 × 135 或 132,013764153662
E-mail:xiemufeng@sina.com.cn
1.1 藥物療效的評價
藥品療效優良的評價標準為:患有該疾病的任何(無論性別、年齡、體質、體內環境)患者服藥后均有一定的療效和作用,即治療有效性好和適用范圍廣。否則就會導致:由不同廠家生產的相同劑型的同一藥品,對不同患者有不同的療效;同一廠家的同一藥品對不同患者療效不同,即治療有效性低、范圍窄。
療效的優劣與生物利用度緊密相關。生物利用度低的藥品可能只在某一體內環境 (如胃酸正常者) 才有一定的崩解、溶出和吸收,而對其它患者療效不顯著。這正是藥品內在質量差異的核心所在。
1.2溶出度試驗及評價標準
雖然生物利用度的高低最終是以臨床效果來衡量的,但在很大程度上可依賴體外溶出度試驗來評價。體外溶出度試驗是指在規定介質中,在一定試驗條件下,藥物從片劑或膠囊劑等固體劑型溶出的速度和程度。自1967 年由美國率先推出后,得到迅速推廣,現已成為制劑質量控制的重要衡量指標,成為評價制劑處方和生產工藝極其重要的手段。
提高體外溶出度試驗與體內生物利用度的相關性,及確立溶出度試驗條件來科學有效地進行評價制劑質量是研究的重點之一。
溶出度試驗裝置中的轉籃、槳板及轉速可用于模擬人體胃部和小腸的蠕動。而人體內的消化液,目前國際上通常采用以下 4 種溶出介質來模擬:
pH 1.2 的溶液( 氯化鈉 2.0g,加水適量溶解,加鹽酸7ml,再加水稀釋至1000ml,即得)。國外目前傾向于此配制方法,不同于我國目前通常采用的0.1mol/L 鹽酸液(鹽酸9ml → 1000ml)。
pH 4 乙酸鹽緩沖液0.05mol/L乙酸-0.05mol/L 乙酸鈉(16.4∶3.6)。其中的離子濃度較我國藥典附錄中記載的低。目前我國有關該介質條件下的溶出試驗研究進行得較少。
(2) pH 6.8磷酸鹽緩沖液(磷酸二氫鉀3.4g和無水磷酸氫二鈉3.55g,加水適量溶解并定容至1000ml, 再稀釋一倍,即得) 。其中的離子濃度也較我國藥典附錄中記載的低。
(3) 水一個優質藥品,在采用一定的溶出裝置和轉速時(這些條件也需進行詳盡的研究和論證),在以上4種溶出介質中均應有一定的溶出曲線,這樣就能保證該藥品用于人體時,可在各種體內環境中均有一定的溶出或釋放,即對于任何體質的患者均有一定的療效。如將 4 條(或多條)曲線結合起來,還可用時間、溶出量和溶出介質3三維圖(見圖2)來表示, 應當是平滑、有一定坡度的山坡型。
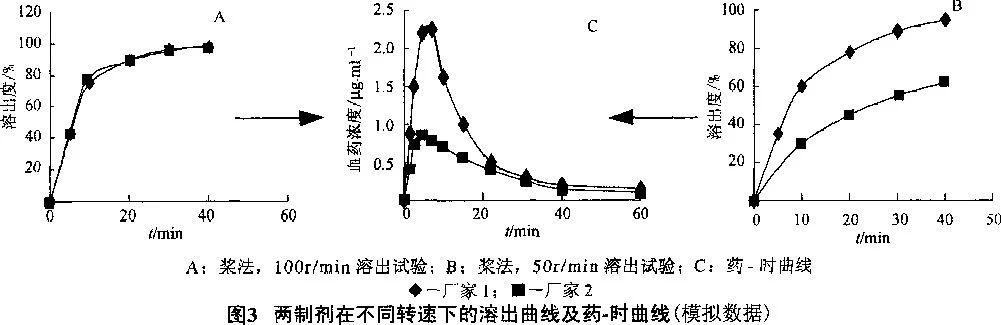
而如果該制劑僅在pH 1.2 條件下溶出較好,就 只能保證胃酸正常的患者吸收良好,而胃酸缺乏的患者可能就較差。人體胃腸道的蠕動程度,個體差異較大。藥物溶出度試驗中,如果該制劑僅能在槳法、100r/min 條件下溶出,那么它也許只在身體機能強壯者的體內釋放和被吸收,而在虛弱者體內,便不能釋放和被吸收;而如果在槳法、50r/min 條件下,在上述 4 種溶出介質中均有“較高的、一定的”溶出曲線,那么無論患者體內情況如何,均會具有較高的、一定的生物利用度,即具有廣泛的療效性。見圖3。
以卡馬西平片為例,日本“醫療用醫薬品品質情報集” 日本厚生省,即日本的參比制劑目錄 (Orange Book),簡稱:參比目錄 中規定該制劑(規格 100mg,卡馬西平不溶于水)在上述 4 種介質中的溶出度試驗條件均為槳法、轉速 75r/min,溶出介質 900ml,分別于 5min 和 30min 取樣測定,限度要求分別為不得大于60%(防止突釋)和不得少于70%。由于我國沒有參比制劑目錄,參照中國藥典
2005 年版二部卡馬西平片項下的方法:采用第二法(槳法)、轉速 150r/min,0.1mol/L 鹽酸 1000ml 為溶出介質,60 min 取樣測定,限度為65 %。筆者曾將某一國產卡馬西平片按照日本參比目錄中的要求進行試驗,結果 4 條曲線與日本參比目錄中所報道的溶出曲線均相差甚遠。
溶出度試驗條件、溶出介質的選擇和限度擬定的差異,勢必導致對處方篩選、制劑生產工藝的不同要求。溶出度試驗條件制訂得嚴格,可促使制劑工藝的改進和提高。
2.日本“藥品品質再評價”( 薬品品質再評価) 工程簡介
日本在藥品臨床使用過程中,也曾出現過以下問題。
(1)如何使市場上不同廠家生產的同一品種藥品對于任何患者均具有相同的生物等效性?
(2)) 如何更為科學、有效地利用體外溶出度試驗來評價或替代體內生物利用度?
(3)如何通過制訂科學、合理的溶出度試驗條件和方法,提高生物利用度?即如何去促進生產制劑工藝的提高與改良,從而完善藥品內在品質、提高療效?
(4)如何能夠更有效、便捷地去保證眾多的仿制藥品具有與原創廠家產品相同品質和臨床療效? (5)如何使后期大批量生產的藥品與臨床試驗時
中試生產的藥品具有相同品質和生物利用度?
(6) 如何保證高通量、高水溶解性的固體制劑藥品在仿制時,減免生物利用度試驗或臨床試驗? 為此,日本厚生省藥品管理部門于1998 年開展了“藥品品質再評價”工程。
2.1 1 “藥品品質再評價”工程的宗旨和技術核心該工程的目的是保證口服固體制劑對不同患者
均有較高的生物利用度;使不同企業生產的同一藥品均有相同的生物利用度;通過現有的技術手段, 對藥品內在品質和有效性重新進行評估。內容是: 通過全面、細致、深入的體外溶出度試驗研究和測定,來評判藥品質量;通過“在嚴格的溶出度試驗條件下,在各種介質中均具有較高的、一定的溶出曲線”這一要求,提高體外溶出度試驗與體內生物利用度的相關性,逐步替代或減少體內生物利用度試驗;推動藥品生產企業對制劑工藝的充分、詳盡研究,進而提高生物利用度試驗和臨床試驗的成功率,或減免生物利用度試驗和臨床試驗 4 ,5 。
溶出度試驗可有效區分同一制劑生物利用度的差異,關鍵是如何確立溶出度試驗的條件?
由此,日本國家藥品審評部門成立了專家小組,確定了“藥品品質再評價”工程的實施方案和操作步驟,并將每一品種的溶出度試驗要求與結果匯編成冊(主要是難溶性藥物),編制成《參比目錄》, 逐批公布,供日本藥品生產企業參照執行。
2.2 2 “藥品品質再評價”工程操作流程
2.2.1溶出試驗條件的規定
以上4 種溶出介質中,采用槳法、轉速50r/min、
溶出介質 900ml,并可根據實際情況適當改變介質pH、轉速或添加表面活性劑(不允許添加有機溶劑) 進行試驗。普通制劑測定時間點分別為 5、10、15、30、45 、60 、90、120min,此后每隔 1h 直至 6 h 止;緩釋制劑測定時間點則分別為15 、30 、45 、60 、9 0 、1 2 0 m i n ,3 、4 、5 、6 、8 、1 0 、1 2 、24h,連續兩點的累積釋放率達95%以上,則可提前結束。
2.2.2 確定品種并進行預試驗
首先確定一批藥物名單( 每批 20 ~30 個),于《參比目錄》專門的網站上公布。然后由生產有關藥品的原創廠家測定本廠產品在以上 4 種溶出介質中的溶出曲線,報送專家小組。小組與原創廠家進行交流溝通后確定 4 條“標準溶出曲線”,于網站上公布。仿制廠家則根據該條件測定本廠生產的相同品種在上述 4 種溶出介質中的溶出曲線,并進行比較后報送專家小組。如一致,則通過;如達不到,則給予適當的期限,進行制劑工藝的改進;如仍不能,則取消該廠生產該藥品的批文。因為不同“坡度”的溶出曲線的生物利用度也不同,因此,所謂的“一致” 并非簡單的目測,而是采用相似因子法(50 ≤ F2 ≤ 100)和Chow's 法6 確定。
2.2.3 結果評判
(1)) 專家小組將委派地方藥品檢驗機構進行抽查,核實溶出度試驗數據。
(2)如仿制廠家能做出更好的溶出曲線或溶出條件,專家小組將會根據原創廠家產品的生物利用度和臨床使用效果,考慮是否將該仿制藥廠的產品替換原創廠家的標準制劑作為參比制劑;如原創廠家藥品的臨床使用效果已很好,則將原創廠家和該仿制廠家的產品同時列入《參比目錄》。
(3)) 如必要,除以上4 種溶出介質外,還可增加pH 為 2.0、3.0、5.0 等溶出介質中的溶出度測定和比較。
( 4 ) 溶出度試驗結果不應受儀器誤差的影響, 即應有良好的耐受性。如出現差異,則說明該制劑工藝尚不成熟,仍需改進。
(5)溶出度試驗應盡量選取弱條件(如槳法、轉速50r/min)進行制劑處方篩選或兩制劑之間的對比試驗,而不應選取強參數(如槳法、轉速 100r/min)。
2.3 3 公布《參比目錄》
在該目錄中,將公布出:有效成分、制劑類型、制劑規格、參比制劑的生產廠家、溶出試驗條件、4 條標準溶出曲線、該制劑的溶出度試驗質量標準、該原料藥的物理化學性質( 主要有解離常數,在 4 種溶出介質中的溶解度,在水、不同p H 溶液和光照條件下的溶液穩定性等)。
2.4 意義和影響
該項工程促進了日本制藥行業對制劑工藝的全面深入研究和嚴格把關,提高了藥品內在品質,也為日本制藥企業占領國際市場起到了促進、推動作用。許多企業均以參比制劑多、品種能收入《參比目錄》而自豪。 同時對中小企業帶來了很大的沖擊,對藥品生產企業的優勝劣汰起到了積極的促進作用。也消除了人們“仿制藥不如原創藥”的想法,認同了仿制藥的品質。 日本是采用藥品由國家定價、同一制劑的價格在全國一致的原則,但政府允許原創藥或參比制劑生產廠家的產品價格可高出同類仿制產品10%~20%。 此外,由于溶出度試驗工作量的大幅增加,使自動溶出儀的市場需求非常旺盛,也間接地促進了藥品檢測儀器的發展和普及。 2.5上市產品的品質保障
為確保后期大批量生產的藥品與臨床試驗藥品具有相同的品質,日本藥品認證部門要求生產企業內控標準應嚴格按照 4 條溶出曲線的要求檢驗樣品(一般情況下,企業僅檢測最難溶出的那條曲線);地方藥檢所不定期進行抽檢。 由此可見,日本藥品審評部門針對每一個( 難溶) 藥物的固體制劑,制定了詳細、統一、科學的溶出度試驗條件和溶出限度要求,避免了藥品仿制時的“低水平重復”,鼓勵制藥企業對制劑工藝進行深入研究,使藥物制劑相關專業人才供不應求; 也拉動了藥用輔料、制藥機械設備等行業的發展。 3 我國現狀
目前我國的一個未被藥典收載的品種,可能有幾十個廠家生產,就會有幾十個不同的質量標準。 溶出度試驗條件有槳板法、轉速 50 ~100r/ min 不等,更有添加表面活性劑甚至有機溶劑的。 研制單位在研發時,往往選用國內某藥廠產品作為參比制劑,有的即使選用國外制劑作為參比制劑,但溶出條件選擇過于寬松,且通常也只比較一種溶出介質,極少比較多種介質。 這就容易造成一種傾向,誤認為溶出度試驗是孤立存在的,很少考慮與體內生物利用度的相關性。目前申報的新藥和仿制藥,以及學術期刊上發表的 有關固體制劑溶出度的研究文章,不少均進入了該 “誤區”。也導致了目前我國制劑工藝的粗糙和低水平重復,也間接挫傷了深入研究制劑工藝、擬定嚴格 溶出試驗條件的企業的積極性。而當藥品收入藥典 或標準轉正時,質量標準擬定時過多照顧各產品的 實際情況,使大部分廠家生產的產品均能夠符合規 定的溶出度試驗條件。 我國許多企業已通過了GMP 認證,而制劑工藝篩選和溶出度試驗條件擬定等是一個專業知識、技術的研究,是屬于“軟件”范疇的。因此不能簡單地以是否通過GMP 認證來評判企業及其產品。 目前仿制藥申報的數目極其龐大,藥品審評中心也任務繁重。如何客觀地評價其內在品質是否與進口制劑生物等效,是值得我們深思的。 國內做生物利用度試驗,均選擇年輕健康者, 結果也比較容易一致。但如果選擇不同人群,情況就可能不同。 4 建議和展望
文獻7指出:應盡快建立我國的“藥品制劑參比目錄”,并應有詳細的參照細則,不應隨意選擇一個藥廠生產的產品或溶出度試驗條件進行比較,因為隨著規定的80%誤差范圍傳遞,最終產品可能偏離很遠。國家食品藥品監督管理局也已從2004 年開始實施“國家藥品標準提高行動計劃”,在隨后的3~ 5 年,將全面清理標準低、質量不可控的藥品,從源頭上著手保證上市藥品的有效性。 4.1 具體建議
“國家藥品標準提高行動計劃”可與建立我國自己的參比制劑目錄緊密地聯系起來。
(1)) 成立專家小組,組建我國的《藥品參比制劑目錄》委員會。由委員會確定工作方針、指導原則、溶出度測定技術細節、方法認證原則等一系列文件,并建立專門的網站,公布相應的計劃和實施的具體步驟,讓生產企業有一定準備。
(2)結合參比制劑可獲得途徑的難易,從難溶性藥物、藥典品種、市場需求量大的品種、患者群高的品種中挑選確定第一批藥物名單。逐批公布需評價的藥品品種目錄,依次有序地進行。
(3)參比制劑,可先從國外知名藥廠在國內合資廠生產的品種中確立,先由該廠進行所指定產品的 4 條溶出曲線測定,能達到一定要求后,可暫時將該合資廠的品種確定為參比制劑。合資廠如不生產參比制劑,可以進口制劑或國內首家能夠達到溶出要求的生產廠家產品,定為我國的參比制劑。必要時,也可考慮參考日本的參比制劑目錄所登載的
(4)在網站上公布第一批藥品名單目錄和相應的“4 條溶出曲線數據”及參比制劑生產廠家,書面通知生產這些品種的所有廠家在一定期限內(如3~ 6 個月) 重新評估本廠產品的溶出度試驗條件及方法,改進制劑工藝以達到目錄中溶出度曲線的要求, 最后報送地方藥檢所進行復核。如生產企業在一定期限內達不到要求,則可根據實際情況,延緩期限, 如仍達不到,則取消該品種生產批準文號。
(5)市場抽查,應主要以溶出度測定為主,且應測定在 4 種溶出介質中的溶出情況。
4.2展望
通過國家藥品審評部門加大對溶出度試驗的審評力度并逐步提高標準,與國際接軌,可促進我國 藥品生產工藝的提高,推動藥品生產企業對制劑工藝的深入研究,帶動藥學高等教育的發展,同時拉 動我國藥用輔料行業、制藥機械設備等行業的發展 和進步,并對藥品研發公司的整合、企業兼并的市 場行為,也必將具有積極的促進作用。目前開展的 “集中藥品招標采購工作”中,建議對多個廠家生產的同類品種進行科學、有效的體外溶出度試驗測定, 對溶出曲線和溶出度良好的藥品,應該成為中標的 首選,使招標工作更易于客觀操作,科學指導。 總之,溶出度試驗是固體藥物制劑的“靈魂”,抓住了這一點,就可“撬動”整個制藥行業和相關產業的全面發展。
致謝:所有對本文提出過寶貴意見和建議的同仁們。
參考文獻: [1]翟發林, 丁青龍, 殷 強. 舒必利片溶出度考察[J].中國醫院藥學雜志, 1991, 11(5): 209-211. [2]Morihara M, Aoyagi N, Kaniwa N, et al. Assessment of gastric acidity of Japanese subjects over the last 15 years[J]. Biol Pharm Bull , 2001, 24(3): 313-315. [3]薛大全, 高鴻慈, 張先洲. 實用片劑制備指南[M]. 湖北: 武漢出版社, 2000. 329-330. [4]日本厚生省醫薬安全局審査管理課. 後発醫薬品の生物學的同等性試験 ンについて[M]. 日本醫薬審第487 號, 1997-12-22. [5]日本厚生省醫薬安全局審査管理課. 仿制藥品的生物等效性研究指導原則醫藥[M]. 第487號, 1997-12-22. [6]夏錦輝, 劉昌孝. 固體藥物制劑的體外溶出度的統計學評價分析[M]. 中國藥學雜志, 2000, 35(2): 130. [7]陳曉媛. 從生物等效性研究中存在的問題談如何提高我國已有國家標準藥品的質量[N]. 中國醫藥報, 2004-01-08.






