

時間:

翻譯:工業藥劑發燒友
審核:華溶應用中心
一、摘要
超級崩解劑是速釋制劑中用于促進片劑快速崩解的關鍵輔料,因此了解超級崩解劑差異對產品性能的影響非常重要。目前的研究使用表面溶出紫外成像考察了超級崩解劑關鍵材料屬性(羧甲淀粉鈉(SSG)的粘度、交聯羧甲基纖維素鈉(CCS)的粒度分布(PSD))對其性能(溶脹)和藥物溶出的影響。采用酸性和堿性介質評價不同pH值對超級崩解劑性能的影響及其對藥物溶出的影響。使用高溶性(撲熱息痛)和難溶性(卡馬西平)藥物作為模型化合物,制備藥物壓片和藥物-崩解劑壓片用于溶出實驗。在與溶出介質接觸時,由于崩解劑的快速水合能力,致密表面上存在溶脹的SSG或CCS層。超級崩解劑的溶脹行為取決于崩解劑的關鍵材料屬性和介質的pH值。由于改善了潤濕或崩解,與不使用超級崩解劑相比,藥物溶出更快。隨著SSG粘度或CCS粒徑的增加,藥物溶出的改善不太明顯。與酸性條件相比,在研究的超級崩解劑存在的情況下,高溶性藥物在堿性條件下的溶出稍迅速,這是由于崩解劑增加水合能力,并且藥物通過膨脹的崩解劑結構快速釋放。對于難溶性藥物觀察到相反的情況。多元數據分析的使用揭示了崩解劑對藥物溶出度的影響。
二、介紹
關鍵材料屬性的確定和控制對于藥物的生產與質量的QBD理念很重要。口服固體劑型中超級崩解劑的存在對于片劑快速崩解和改善藥物溶出至關重要。羧甲淀粉鈉(SSG)和交聯羧甲基纖維素鈉(CCS)(分別為淀粉和纖維素的聚合物)通常用作片劑制造中的超級崩解劑。超級崩解劑促進片劑崩解的主要機制是溶脹(Quodbach和Kleinebudd,2016)。膨脹是指超級崩解劑顆粒與水接觸時的體積膨脹(Quodbach和Kleinebudd,2016)。SSG和CCS是鈉鹽,在酸性條件下以中性形式存在,在堿性條件下以離子化形式存在。它們源自天然聚合物,經過天然聚合物鏈的兩個主要修飾步驟(羧甲基化和交聯化),以提高崩解劑功能(Quodbach和Kleinebudd,2016)。首先,羧甲基化增加了聚合物的親水性,并允許水進入崩解劑(Zhao和Augsburger,2006)。其次,由于天然聚合物是部分可溶的,其溶出可能會增加介質的粘度,聚合物鏈的交聯有助于降低聚合物的可溶含量。SSG通過磷酸基團交聯(Edge和Miller,2005),而CCS通過酯基團交聯(Guest,2005)(圖1)。與天然聚合物相比,SSG和CCS作為片劑崩解劑的優異性能歸因于這兩個改性步驟。試驗證明了與水接觸后SSG和CCS的變大和快速膨脹,SSG和CCS的體積中值直徑(平均體積尺寸)增加(SSG與水接觸時為123μm,干粉為35μm;CCS接觸水時為92μm,而干粉為45μm)和液體吸水量增加,(SSG和CCS在120秒內吸水分別為16g/g和10g/g)Zhao和Augsburger,2005)。盡管SSG和CCS的結構相似,但是它們的膨脹(SSG的三維膨脹,CCS的二維膨脹)存在差異(Rojas等人,2012;Zhao和Augsburger,2005)和它們的不同交聯方式(SSG中的磷酸基團與CCS中的酯基團相比,聚合物鏈之間的間距更大)(Rojar等人,2012)。分子特性(取代度、交聯度)、顆粒性質(粒度分布(PSD))和水平已被確定為SSG和CCS影響產品性能的潛在關鍵材料屬性(Zarmpi等人,2017)。增加SSG和CCS的取代度會導致更快的吸水率和溶脹,但需要確定最佳值,因為高羧甲基化程度可能會導致介質粘度增加(Zarnpi等人,2017)。據報道,當增加SSG和CCS的交聯度、粒徑或水平時,會出現更大的溶脹和更快的崩解(Zarmpi等人,2017)。 目前評估超級崩解劑性能和差異的方法包括測定:制劑的崩解時間、粉末/片劑的吸水量、超級崩解劑的膨脹體積、片劑崩解過程中的力(片劑內部的崩解力必須超過粘附力)、藥物的溶出速率、以及片劑崩解后產生的顆粒的大小(Quodbach和Kleinebudd,2016)。實時表面溶出紫外成像目前用于制藥領域,提供了關于崩解/溶出現象的特別信息。紫外溶出成像在QbD方法中很有價值,因為它提供了對藥物溶出初始階段表面狀態的機理理解和早期藥物發現中新藥候選的表征(Kuentz,2015;Niederquell和Kuentz)。該技術利用集成有UV-vis攝像機和小型流通池,以及一個注入層流下溶出介質的泵(Ostergaard等人,2014)。通過測量流通池的光透射率,在空間和時間上表征溶出物質,并有助于識別藥物固有溶出速率、表面溶脹/崩解/溶出現象、濃度梯度和微環境pH變化(Gordon等人,2013)。通過使用實時表面溶出成像,對原料藥(Ostergaard等人,2014)、崩解劑(Pajander等人,2012)及其相互作用(Colombo等人,2015;Hiew等人,2018)的溶出行為進行了深入研究。
超級崩解劑的存在或差異對生物藥性能的影響尚不清楚。胃腸道因素可能影響超級崩解劑的性能,因為pH值是最有影響的SSG和CCS的電離模式。酸性輔料在酸性介質中的水合能力低于在堿性介質中電離狀態的水合能力(酸性和堿性介質是根據生理pH范圍定義的(Sjogren等人,2014)),這導致酸性條件下的膨脹減少(Zhao和Augsburger,2005)。SSG在水中和pH =1的0.1M HCl中的體積增加百分比分別為251%和43%,CCS分別為104%和51%(Zhao和Augsburger,2005)。超級崩解劑對產品性能的影響也與藥物性質有關。陽離子藥物與CCS羧基的相互作用會影響常規藥物分析。在樣品處理過程中,含有CCS的片劑配方可能會損失活性藥物成分(API),因為據報道,由于帶電藥物-崩解劑相互作用,存在CCS的溶液中藥物(二甲雙胍(Huang等人,2006)、依司他普侖(Larsen和Melander,2012))的回收率較低。陽離子藥物從含有SSG和CCS的速釋片劑中溶出的延遲也歸因于帶電藥物-輔料相互作用(Balasubramaniam等人,2008)。
目前評估超級崩解劑性能和差異的方法包括測定:制劑的崩解時間、粉末/片劑的吸水量、超級崩解劑的膨脹體積、片劑崩解過程中的力(片劑內部的崩解力必須超過粘附力)、藥物的溶出速率、以及片劑崩解后產生的顆粒的大小(Quodbach和Kleinebudd,2016)。實時表面溶出紫外成像目前用于制藥領域,提供了關于崩解/溶出現象的特別信息。紫外溶出成像在QbD方法中很有價值,因為它提供了對藥物溶出初始階段表面狀態的機理理解和早期藥物發現中新藥候選的表征(Kuentz,2015;Niederquell和Kuentz)。該技術利用集成有UV-vis攝像機和小型流通池,以及一個注入層流下溶出介質的泵(Ostergaard等人,2014)。通過測量流通池的光透射率,在空間和時間上表征溶出物質,并有助于識別藥物固有溶出速率、表面溶脹/崩解/溶出現象、濃度梯度和微環境pH變化(Gordon等人,2013)。通過使用實時表面溶出成像,對原料藥(Ostergaard等人,2014)、崩解劑(Pajander等人,2012)及其相互作用(Colombo等人,2015;Hiew等人,2018)的溶出行為進行了深入研究。
本研究目的是評估具有不同關鍵材料屬性的崩解劑的崩解性能以及崩解劑的差異對藥物溶出的影響和作用。使用實時表面溶出紫外成像進行崩解劑表征和崩解劑存在與否時的藥物溶出情況研究。通過選擇三種不同粘度類型的SSG和兩種不同PSD的CCS,研究了輔料差異對輔料溶脹和藥物溶出的影響。使用高溶性撲熱息痛;BCSIII類(Kalantzi等人,2006)和難溶性(卡馬西平;BCS II類(Kovacevic等人,2009))藥物來評估崩解劑差異和藥物特性對藥物溶出的相互作用。在酸性和堿性介質中進行研究,以評估pH值對超級崩解劑溶脹和藥物溶出的作用。
三、材料與方法
3.1 材料
原料藥:對乙酰氨基酚(PRC,I型)從Fischer Scientific(英國)獲得。卡馬西平(CBZ,III型)購自Fagron(英國)。輔料:SSG品牌:Glycolys LV低粘度(60分鐘時水溶液的粘度=10.8cP)和Glycolys高粘度(60 min時水溶液粘度=20.9cP))(Roquette,法國)、Explotab CLV低粘度(60分鐘時水溶液的粘度=12.7cP)(JRS Pharma,USA)以及CCS品牌:AcDiSol低粒徑(dso=74.2 um)(FMC,USA)、Primellose高粒徑(do=109.8 um)(DFE Pharma,Germany) 。化學品:鹽酸36.5%-38%,HPLC級甲醇從Sigma Aldrich(英國)獲得。氯化鈉、氫氧化鈉、磷酸二氫鉀購自Fisher Scientific(英國)。水為超純(Milli-Q)實驗室級。過濾器:聚四氟乙烯(PTFE)13 mm過濾器0.45μm孔徑購自Fisher Scientific(英國)。
3.2 儀器
賽托利斯BP 210 D天平(英國Sartorius有限公司)、梅特勒-托利多Seven Compact S210型 pH計(瑞士梅特勒·托萊多)、VortexGenie 2渦旋混合器(美國科學工業公司)、安捷倫科技1100系列HPLC(四元泵(G1311A)、自動采樣器(G1313A)、柱溫箱(G1316A)、二極管陣列檢測器(G1329A)和Chemstation軟件(美國安捷倫科技公司)、Actipix SDI300溶出成像系統(英國Paraytec有限公司)與流通池(推薦華溶DS-7CP流池法溶出系統)、Quickset小扭矩螺絲刀(英國Torqueleader)、Actipress 316不銹鋼壓力機(英國Paraytec LtD)。
3.3 方法
3.3.1 體外溶出介質
根據歐洲藥典(pH Fur 2014)中描述的方法制備介質( pH=1的0.1MHCl、pH 6.8磷酸鹽緩沖液)
3.3.2 壓片
對于崩解劑表征,將20mg每種崩解劑倒入片劑中(不銹鋼圓柱體,內徑2mm,高度:2.4mm),并使用手動壓片機以75cNm的恒定扭矩壓實5分鐘(Pajander等人,2012)。為了進行溶出研究,制備了純原料藥(撲熱息痛(PRC)、卡馬西平(CBZ))的壓片以及原料藥-超級崩解劑的壓片。將10 mg API(PRC,CBZ)倒入片劑中,并使用手動壓片機在75 cNm的恒定扭矩下制備純API壓塊(藥物壓塊)5分鐘(Pajander等人,2012)。使用手動壓片機在75 cNm的恒定扭矩下制備具有API-超級崩解劑壓塊(藥物崩解劑壓塊),持續5分鐘(Pajander等人,2012)。
3.3.3 體外實時表面溶出紫外成像
使用Actipix SDI300表面溶出成像系統和流通池進行實時表面溶出UV成像。流通池由裝有石英池(7 mm高、4 mm寬、62 mm長)和聚醚醚酮(PEEK)樣品架的濾筒組成。光源是脈沖氙燈,帶濾波器(檢測波長±10nm)用于選擇適宜的波長。溶出介質通過注射泵注入池內。溫度控制單元用于保持恒定溫度。紫外成像儀的檢測面積為9mm x 7mm(1280像素x1024像素),像素大小為7um x 7um。儀器的詳細描述已在之前介紹(Longet等人,2019年:Ostergaard等人,2014年)。
對于崩解劑的表征,在254nm下,在37°C、pH=1的0.1MHCl和pH為6.8的磷酸鹽緩沖液中,在靜止條件進行5分鐘的實驗。在280nm下,使用1mL/min的流速在37℃、pH=1的0.1MHCl和pH6.8的磷酸鹽緩沖液中進行藥物壓片和藥物-輔料壓片的體外藥物溶出實驗20分鐘。對于兩種崩解劑表征實驗和體外藥物溶出研究,在流通池中充滿溶出介質,沒有藥片時,記錄了黑暗(燈關閉時持續10s)和標準狀態(燈打開時持續10秒)的圖像。開始數據收集,60秒后暫停數據記錄,并將藥片引入池內。用溶出介質沖洗系統,以避免流通池中出現氣泡,并恢復數據采集。使用Actipix D100軟件,版本1.8.50805(英國Paraytec有限公司)將指定量化區域內的像素強度轉換為吸光度值。在體外藥物溶出實驗中,在UV圖像的量化區域中存在溶脹崩解劑(SSG和CCS品牌)導致光的散射或物理阻擋的增加(Long等人,2019)。在這些情況下,每隔1分鐘收集洗脫樣品,通過PTFE 0.45μm孔徑過濾器過濾流出物樣品,并通過HPLC進行分析。之前對每種藥物進行了一式三份的過濾器吸附研究,并確認所研究藥物在所用過濾器上沒有吸附問題。所有實驗一式三份。
3.3.4 色譜條件
通過HPLC分析溶出樣品(流出物收集)。HPLC分析程序是對PRC(Gao等人,2014)和CBZ(Vertzoni等人,2006)已發表方法的修改。兩種藥物均使用反相Spherisorb(Waters)C18柱(250 x 4.6 mm,5μm)。對于PRC,流動相為甲醇和水20:80(v/v),溫度保持在20℃。進樣體積為20μL,檢測波長為257nm。對于CBZ,流動相由甲醇和水60:40(v/v)組成,溫度保持在25℃。進樣體積為100μL,檢測波長為285nm。兩種藥物的流速均設定為1mL/min(等度)。PRC和CBZ的洗脫時間分別為6分鐘和4分鐘。根據標準曲線進行藥物定量。標準品由溶于甲醇(PRC:2mg/mL,CBZ:1mg/mL)的藥物濃縮儲備溶液制備。PRC和CBZ的校準曲線范圍分別為10-300μg/mL和0.5-50μg/mL。
3.3.5 體外溶出數據處理
對于溶脹超級崩解劑行為的表征,由于i.所研究聚合物的不溶性和ii.無法獲得崩解劑濃度梯度的定量數據,記錄的高吸光度值可能歸因于溶脹聚合物對光的吸收或散射,或未溶出聚合物顆粒對光的物理阻擋(Pajander等人,2012)。通過吸光度作為離片劑中心的距離的函數,只能獲得溶脹速率和程度的定性信息。使用Actipix D100軟件版本1.8.50805(英國Paraytec有限公司)根據像素強度自動計算吸光度值(Abs)(圖像區域尺寸:4.6 mm×1.3 mm)。使用SigmaPlot 13.0描述所研究SSG和CCS在 pH=1的0.1MHCl和pH 6.8的磷酸鹽緩沖液中的溶脹行為(吸光度值作為與片劑中心距離的函數)的分類梯度圖(其中z變量沿x和y方向的變化通過顏色變化來說明)(美國Systat軟件公司)。根據樣品中測得的藥物濃度(基于HPLC分析數據)和片劑中的藥物量計算藥物溶出的累積百分比。構建了作為時間函數的藥物累積溶出百分比的溶出曲線。根據樣品中測得的藥物濃度(基于HPLC分析數據)和溶出實驗的已知流速,計算實驗期間每1分鐘間隔的藥物溶出速率(ug/min)。繪制了描述藥物溶出速率隨時間變化的曲線圖(間隔1分鐘),并在采樣間隔的中間點顯示了溶出率的標準偏差(SD)。使用梯形法計算的截至最后實驗時間(20分鐘)的溶出曲線下面積(AUC)用于表征藥物溶出。根據E.(1)計算每種超級崩解劑對藥物溶出的相對影響(RE)。
其中AUCC和AUCT分別是對照和試驗的片劑的溶出曲線下的面積。進行了兩組比較:在第一組(第1組)中,檢測了在各個介質里藥物片劑與藥物-輔料壓片片劑的溶出的差異,將藥物壓片和藥物-輔料壓片的溶出曲線的AUC作為對照溶出曲線和測試溶出曲線,在第二組(第2組)中,以酸性和堿性條件下的溶出曲線的AUC分別作為對照溶出曲線和試驗溶出曲線,研究了每個藥物-輔料壓片的片劑在酸性和堿性情況下藥物溶出的差異。輔料對藥物溶出影響的風險評估通過對溶出曲線AUC的RE(設定參考范圍標準-20%-25%)(FDA,2002年)進行評估(選擇該范圍是為了評估口服給藥后藥物暴露的差異;即生物等效性研究)。崩解劑對溶出曲線的AUC的RE超出這些值(RE<-20%或RE>25%)被認為對口服藥物性能可能至關重要。
3.3.6 體外溶出數據的多變量數據分析
通過使用XLSTAT軟件(美國微軟公司)的多元線性回歸(MLR),輔料RE對藥物溶出的影響與輔料關鍵材料屬性(如SSG的粘度、CCS的PSD)、藥物水溶性和介質(酸性、堿性)特性相關。在SSG(模型1)和CCS(模型2)存在下,構建了崩解劑對溶出曲線AUC的RE的兩個模型。兩種模型的因素評估很明確,包括:i.藥物水溶性0:難溶性,1:高溶性;基于化合物的BCS(生物制藥分類系統)分類(高溶性:BCSⅠ類和III類;難溶性:BCSII類和IV類)(FDA,2017)。ii.介質(0:酸性,1:堿性)。iii.輔料特性(0:低崩解特性,1:高崩解性能;SSG黏度大小以及CCS的粒度。使用溶出曲線AUC對應的輔料RE作為響應。所選的交互因素包括每種輔料型號與每種藥物的水溶性和介質特性(酸性、堿性)。根據擬合程度與方差擴大因子(VIF)評估生成的MLR模型。高R2值和VIF值<5表明自變量之間缺乏多重共線性的成功模型(Montgomery和Peck,1992)。標準系數用于表示每個變量的方向(正或負)和程度。通過p值評估變量95%置信區間的顯著性(p<0.05被認為是模型中最顯著的(Montgomery和Peck,1992))。
四、結果與討論
用實時表面溶出紫外成像表征超級崩解劑的溶脹行為。
所研究的崩解劑類型和型號先前已根據SSG的粘度和CCS的PSD進行了表征(Zarmpi等人,2019)。Glycolys LV(10.8 cP)和Explotab CLV(12.7 cP)的水溶液60分鐘后的粘度低于Glycolyss(20.9 cP),因為它們的交聯度更高,可溶性物質含量更低(Shah和Augsburger,2001)。與Primellose(d10:21.8μm,d50:52.2μm,d90:109.8μm)相比,AcDiSol由更小的顆粒組成(d10:12.8μm,d50:31.9μm,D60:74.2μm),因此確定了不同CCS的PSD的差異。
表面溶出UV成像用于研究SSG和CCS在緩沖液中的溶脹性能,崩解劑與溶出介質接觸時的溶脹UV圖像見圖1。所研究的超級崩解劑在 pH= 1的0.1 MHCL條件下的溶脹行為隨時間和距離片劑中心的關系如圖2所示。強信號位置表明存在密集膨脹的聚合物結構(Colombo等,2015),因為記錄的高吸光度歸因于膨脹的超級崩解劑的光散射或未溶出的顆粒對光的物理阻礙(如前文所述)。無論崩解劑型號或種類如何,所有研究的超級崩解劑在距片劑中心1.2 mm的距離處,在約20-40秒的時間內膨脹,證明了崩解劑的快速膨脹。對于SSG,與Glycolys LV和Explotab CLV (Abs1.2-1.6 AU)相比,Glycolys表現出較低的吸光度值(Abs1.0-1.2 AU),這可能是由于高粘度品牌的可溶性含量較高(Shah和Augsburger,2001)。對于CCS,與AcDiSol (Abs1.0-1.2 AU)相比,Primellose (Abs1.4-1.6 AU)的較高吸光度值可歸因于較大顆粒對光的明顯物理阻塞(Van Eerdenbrugh等人。2011)。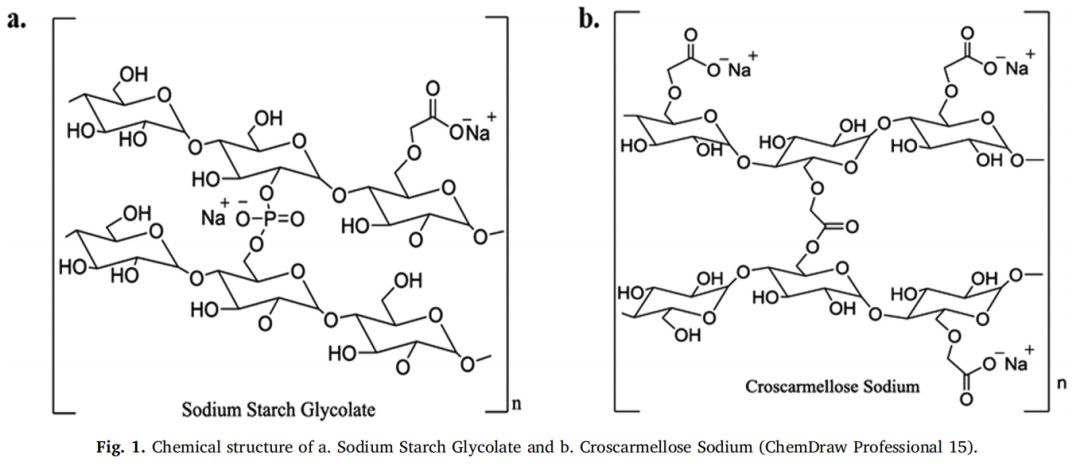
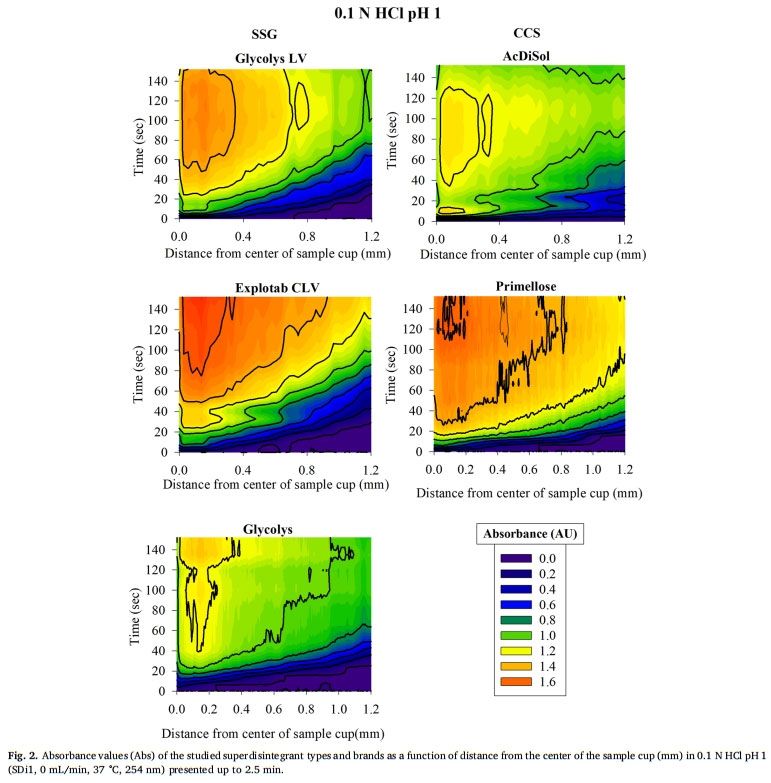
在pH為6.8的磷酸鹽緩沖液中,所研究的超級崩解劑的溶脹行為隨時間和距離片劑中心的函數如圖3所示。與 pH=1的0.1MHCl(20-40 s)相比,觀察到pH 6.8的磷酸鹽緩沖液中的崩解劑溶脹稍快(<20 s),這是由于在堿性介質中崩解劑是電離形式存在(Zhao and Augsburer,2005)以及有較高的液體攝取量(pH=1的0.1MHCl介質:2分鐘后SSG和CCS的液體攝取約為5 g/g;在堿性介質中崩解劑以電離形式溶脹:2分鐘后SSG和CCS的液體攝取分別為18 g/g和10 g/g(Zhao和Augsburger,2005))。在pH為6.8的磷酸鹽緩沖液中的吸光度值(SSG:0.6-1.2 AU,CCS:1.0-1.4 AU)低于pH=1的0.1MHCl介質(SSG:1.0-1.6 AU, CCS:1.0-1.6 AU)。
我們假設,與酸性介質相比,堿性介質中的較低吸光度與磷酸鹽緩沖液pH6.8中崩解劑電離形式的較高膨脹性能有關。因為膨脹聚合物鏈的間距越高(Rojas et al.,2012),光的散射或物理阻擋就越少。在研究的SSG中未觀察到吸光度值的差異(Abs=0.6-1.0AU)。對于CCS,與Primellose(Abs 1.2-1.4 AU)相比,AcDiSol(Abs 1.0-1.2 AU)的吸光度值較低,這是因為其粒徑較低24。與CCS相比,SSG在pH 6.8的磷酸鹽緩沖液中的較低吸收值可以表明SSG的更大的溶脹,這是由于兩種崩解劑交聯類型(SSG為磷酸基,CCS為酯基)的差異以及在與模擬腸液接觸時的膨脹尺寸類型的差異(SSG的三維膨脹(半球狀顆粒)和CCS的二維膨脹(纖維狀顆粒)差異(Rojar等人,2012年)。在研究介質中觀察到的崩解劑性能差異表明,預測胃和小腸之間的崩解差異(由于這兩個內環境的pH值不同),并可能影響產品性能和藥物生物利用度。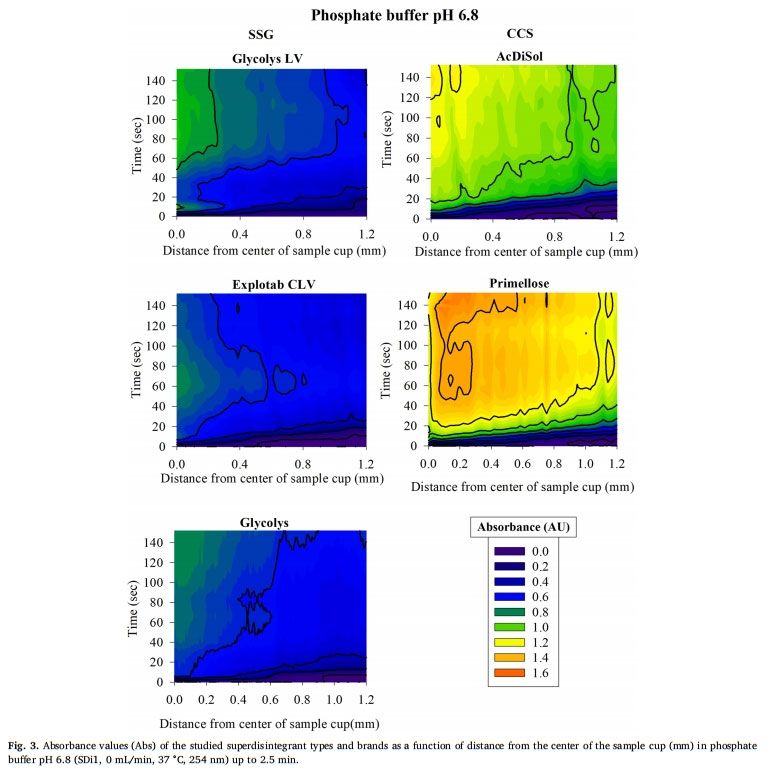
五. 測定SSG差異對藥物溶出的影響
5.1 高溶藥物(PRC)
圖4顯示了在 pH=1的0.1MHCl介質和pH為6.8的磷酸鹽緩沖液中,PRC藥物的壓縮物(對照)和藥物- SSG壓縮物的溶出曲線和溶出速率。在兩種介質中,約5%的PRC在20分鐘內從藥物中溶出。藥物-SSG壓片在20min內溶出的百分比有所增長:在pH=1的0.1MHCl介質中(在Glycolys LV、Explotab CLV和Glycolyss存在下,分別為21%、23%和25%的藥物溶出)和在pH為6.8的磷酸鹽緩沖液中(在Glycolys LV、Explotab CLV和Glyclys存在下,溶出的藥物分別為22%、27%和21%)。在所研究的兩種介質中的實驗期間,PRC從藥物壓塊中的溶出速率都很慢(HCl介質和6.8介質中,20min時溶出速率分別約為20μg/min和17μg/min)。在所研究的介質中,藥物- SSG壓片中的藥物溶出速率比藥物壓片中的藥物溶出速度更快,特別是在早期時間點(1-5 min)。在崩解劑存在下PRC的溶出速率增加,(在兩種介質中5分鐘時,含Glycolys LV、Explotab CLV和Glycolyss的壓塊的溶出速率約為200μg/min)。這可以通過崩解劑快速水合和溶脹(崩解劑在前40秒內在距片劑中心1.2 mm的距離處溶脹)來解釋。這改善了片劑的潤濕(Onuki等人,2018)。與藥物壓塊相比,崩解也可能有助于藥物更快的從藥物-崩解劑壓塊中溶出。與藥物壓塊相比,藥物-崩解劑壓制的片劑更快的崩解有助于藥物更快的溶出,因為在每次實驗結束后,僅在藥物-崩解劑壓片的表面觀察到了顆粒的擴散。溶出曲線的AUC比較表明,與藥物壓片的溶出相比,藥物-崩解劑壓片的藥物溶出更完全(RE>25%)(圖5a)。未觀察到所研究SSG類型在酸性和堿性條件下PRC藥物從藥物-SSG壓塊中溶出的差異(Glycolys LV、Explotab CLV和Glycolyss的RE分別約為5%、20%和20%)。觀察到的微小REs表明,SSG的溶脹性能的pH依賴可能不會顯著影響高溶性藥物在酸性和堿性條件下的溶出。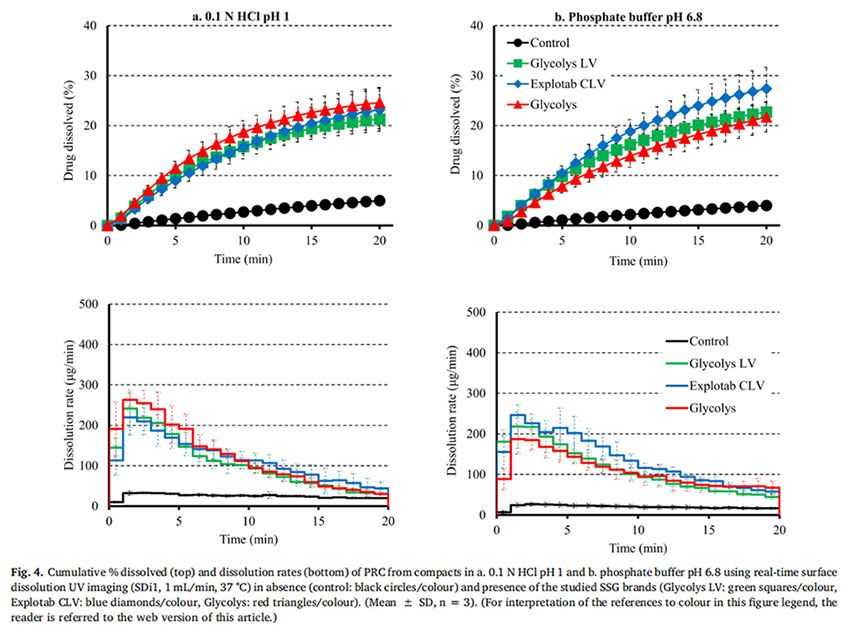
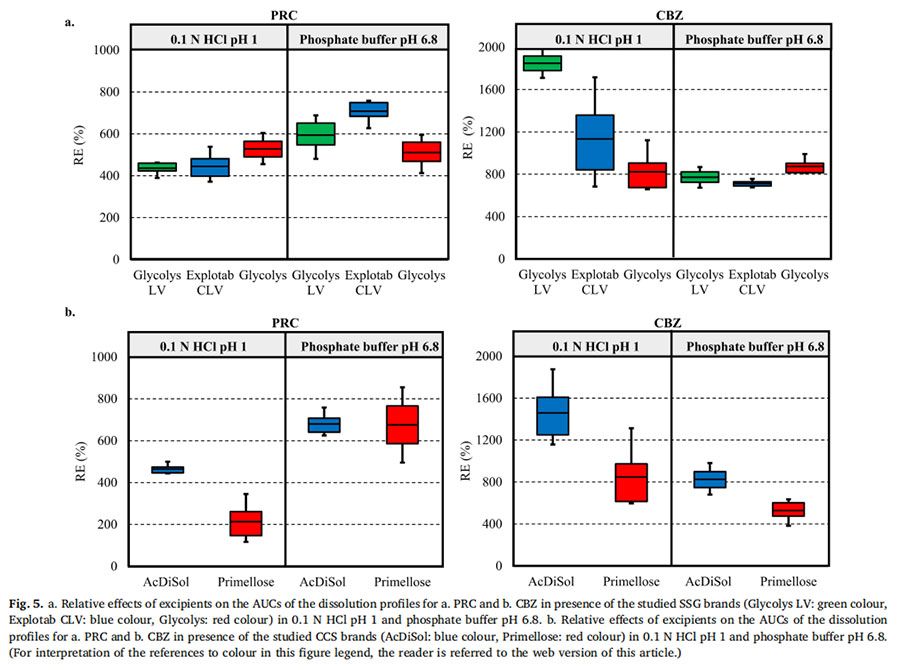
5.2 難溶藥物(CBZ)
CBZ從藥物壓塊與藥物-SSG壓塊在HCl和pH 6.8介質中的溶出曲線和溶出速率如圖6所示。在20分鐘內,HCl和pH 6.8介質中約0.1%的CBZ從藥物壓塊中溶出。在pH=1的HCl中,在Glycolys LV、Explotab CLV和Glycolyss存在下,分別在20分鐘內溶出2.3%、1.6%和1.3%。不同藥物-崩解劑壓片之間,pH 6.8介質、20分鐘CBZ溶出百分比相似(含Glycolys LV、Explotab CLV、Glycolyss的藥物-崩解劑壓片:約1.5%的CBZ溶出)。在兩種介質中,藥物從藥物壓塊中的溶出速度都很慢(在HCl和6.8中,20分鐘時的溶出速度約為0.5 ug/min)。在SSG存在下,CBZ溶出加快,尤其是在早期時間點(1-5分鐘),這可能是由于崩解劑快速膨脹或片劑崩解(如PRC中所解釋的)。在HCl介質中,在Glycolys LV、Explotab CLV和Glycolyss存在下,5min時,CBZ從藥物-崩解劑壓塊中的溶出速率分別為13μg/min、8.6μg/min和6.2μg/min。與低粘度型號(Glycolys LV、Explotab CLV)相比,Glycolyss存在時溶出的藥物量較低,這可能是因為高粘度Glycolyse使壓塊周圍凝膠的粘度增加所致(Quodbach和Kleinebudd,2016)。含低粘度和高粘度SSG品牌的壓塊的CBZ溶出速率的顯著差異在后期逐漸減小。在pH 6.8的磷酸鹽緩沖液中,與沒有崩解劑相比,CBZ的溶出速度加快,三種藥物-崩解劑壓片的溶出速率相似(在所研究的三種SSG存在的情況下,5min時,CBZ溶出速率約為7μg/min)。與無崩解劑相比,有崩解劑的CBZ溶出更完全(RE>25%)(圖5a)。觀察了酸性和堿性條件下藥物-輔料壓片的藥物溶出度差異。對于低粘度崩解劑(Glycolys LV和Explotab CLV),酸性和堿性條件下溶出曲線AUC的RE為負值(Glycoys LV和expltab CLV的RE分別為-45%和-20%),表明與pH=1的HCl介質相比,pH為6.8的磷酸鹽緩沖液中CBZ溶出的改善不明顯。盡管在堿性條件下聚合物吸水更快,由于超級崩解劑的電離作用,在堿性條件下比酸性條件下膨脹得更大(Rojas等人,2012),片劑頂部高度膨脹層的存在可能會對難溶性藥物的釋放增加物理或擴散屏障(Long等人,2019)。溶出曲線的比較表明,在pH 6.8的磷酸鹽緩沖液中的溶出略為完全(REs為30%)。溶出曲線AUC上的正RE可能與Glycolys的凝膠效應(Quodbach和Kleinebudd,2016)以及在pH=1的HCl介質中觀察到的CBZ溶出較慢(與其他兩種崩解劑相比)有關。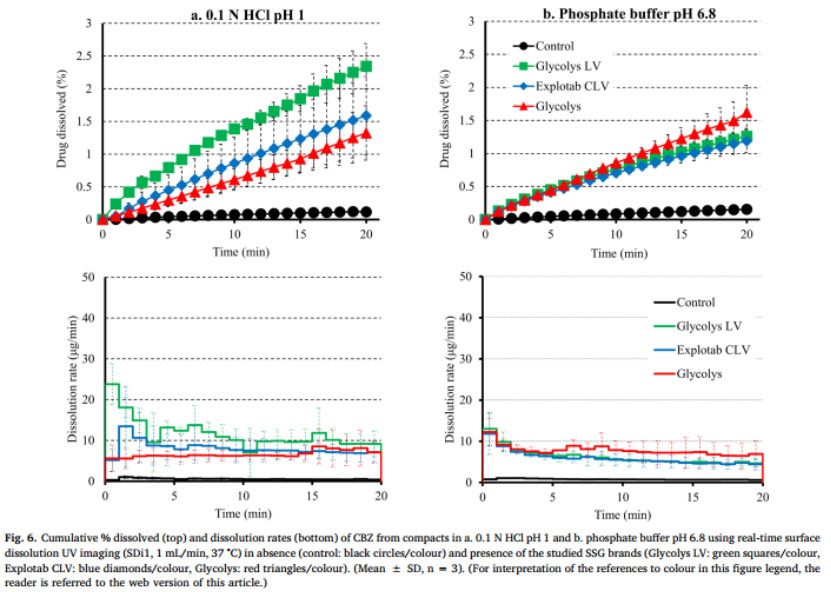
六、 測定CCS差異對藥物溶出的影響
6.1 測定CCS差異對藥物溶出的影響
PRC從藥物壓塊(對照)和藥物-CCS壓塊在pH=1的HCl和pH 6.8磷酸鹽緩沖液中的溶出曲線和溶出速率如圖7所示。與無崩解劑的RPC藥物壓片的溶出相比(20 min:5%),有崩解劑存在時,0.1N HCl(AcDiSol:23%,Primellose:14%)和pH為6.8的磷酸鹽緩沖液(AcDiSol:25%,Primellose:26%)。與在兩種介質中不存在崩解劑相比,崩解劑存在時藥物溶出速度更快,這可能是由于CCS的親水性和溶脹性而導致潤濕或崩解增強所致(Quodbach和Kleinebudd,2016)。在0.1N HCl的早期時間點觀察到所研究藥物-CCS壓塊的溶出速率的差異,因為與AcDiSol(5min:182μg/min)相比,可能是由于在壓塊表面頂部存在較大的崩解劑顆粒(Primellose)。在pH 6.8的磷酸鹽緩沖液中,在含有AcDiSol和Primellose的藥物-CCS壓塊之間觀察到類似的藥物溶出(在兩種CCS型號存在下,5分鐘的溶出速率約為200μg/min)。在后續的時間點,在兩種介質中均存在CCS的情況下,PRC的溶出率逐漸降低,并且所研究的CCS型號之間的藥物溶出差異減小。藥物和藥物-CCS壓片的溶出曲線的AUC比較顯示,含有崩解劑的片劑的溶出明顯更徹底(RE>25%)(圖5b)。如AcDiSol在酸性和堿性條件下的溶出曲線AUC上的RE(RE=10%)所揭示的,未觀察到酸性和堿性狀態下藥物-AcDiSol壓片的藥物溶出差異。與0.1 N HCl介質(RE=97%)相比,在pH 6.8的磷酸鹽緩沖液中,藥物-Primellose壓片的溶出明顯更完全,這可能是因為在0.1 N HCl中存在較大的崩解劑顆粒。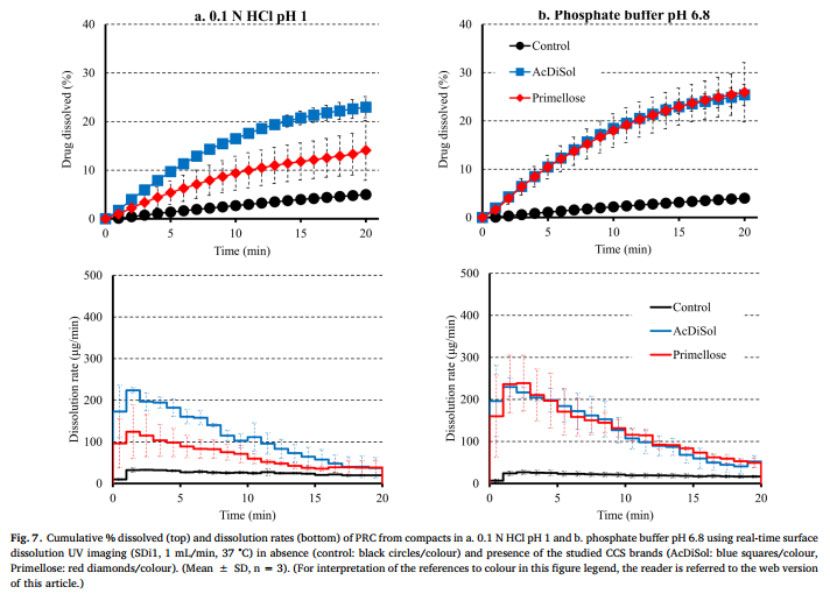
6.2 難溶藥物(PRC)
CBZ在0.1 N HCl 和pH 6.8磷酸鹽緩沖液中從藥物壓片和藥物-CCS壓片中的溶出曲線和溶出速率如圖8所示。含AcDiSol和Primellose的壓塊在0.1N HCl中的CBZ溶出度分別達到約1.8%和1.5%,在pH 6.8磷酸鹽緩沖液中的CBZ溶出度分別為1.3%和1.0%。在溶出早期時間點,崩解劑的快速膨脹導致藥物-CCS壓塊的溶出速度比兩組介質中的藥物壓塊更快。
與 AcDiSol存在下的溶出相比(酸、堿介質溶出分別為:9μg/min與7ug/min),Primellose存在下的溶出偏慢(酸、堿介質溶出分別為:7μg/min and 5ug/min)。可能是由于在壓片的表面的頂部存在較大的崩解劑顆粒(Primellose)。在所研究的崩解劑型號之間的后期時間點的藥物溶出差異在變小。在崩解劑的存在下,在兩種實驗條件下,與對照樣品相比,藥物溶出明顯更完全(RE>25%)(圖5b)。與堿性條件相比,CCS的存在時,CBZ的在酸性條件下溶出增強更為明顯(AcDiSol和Primellose在酸性和堿性條件下的溶出曲線AUC的RE分別為-30%和-20%)。崩解劑在酸性介質中更快的吸水(Rojas等人,2012)預計會導致更快的藥物溶出,這是由于改進的片劑的潤濕性,但這并沒有被觀察到,因為片劑表面頂部的溶脹層可能會產生物理或擴散屏障,延遲藥物釋放(Long等人,2019)。對于不含崩解劑的溶出實驗,在某些情況下,僅在前3分鐘觀察到較高的變異性(變異系數(CV%)>20%),在隨后的時間點觀察到較低的變異性。對于崩解劑存在的溶出實驗,確定了變異性增加的情況(早期和晚期時間點分別為15%
七、 體外溶出數據的多變量數據分析
SSG和CCS模型中的標準系數的變量如圖9所示。兩個模型顯示出良好的擬合(SSG:R2=0.5,CCS:R2=0.5)。對于SSG,崩解劑類型(負相關,p<0.05),藥物(負相關,p<0.05)和介質(負相關,p<0.05)是模型中的關鍵變量。崩解劑類型的負相關表明,與低粘度SSG相比,增加SSG的粘度后溶出改善不明顯。由于低粘度SSG吸水更快,膨脹更大,因此推測會增加片劑的潤濕性(Abraham等人,2016)。高粘度SSG形成的凝膠層會延遲藥物溶出(Quodbach和Kleinebudd,2016)也可以解釋這一現象。不同的SSG粘度對難溶性藥物的影響更為顯著,因為難溶性的藥物將因潤濕性的改善而溶出改善。不同介質的負效應表明,與堿性條件相比,在酸性條件下、SSG的存在時顯著增強了藥物溶出(尤其是對于難溶性藥物)。
對于CCS,崩解劑類型(負效應,p<0.05)和藥物(負作用,p<0.05)是模型中的關鍵變量,增加CCS的粒徑對藥物溶出的改善不太明顯,這可能是由于較大崩解劑顆粒形成了藥物溶出的物理或擴散屏障。CCS的存在改善了潤濕與崩解,促進難溶性藥物的溶出。多變量數據分析表明,CCS差異可能對藥物溶出的初始階段至關重要。
八、總結
超級崩解劑的差異以及互換性對口服固體制劑的性能具有較大影響,因為藥物制劑中超級崩解劑的存在直接影響藥物溶出。在這項研究中,使用表面紫外成像可以對SSG和CCS溶脹及其對藥物出的影響進行半定性分析。證實了SSG和CCS的快速溶脹能力,這取決于關鍵材料屬性和溶出介質的pH。這些結果表明,在口服固體劑型中,SSG和CCS不應被認為是可互換的。超級崩解劑在含有高規格和難溶性化合物的壓塊中的存在導致兩種藥物的溶出速度顯著加快,這可能是由于親水崩解劑增強了片劑的潤濕或崩解。在口服藥物開發中需要仔細考慮具有不同材料屬性的崩解劑的影響,因為不同的崩解劑關鍵材料屬性會影響藥物的溶出程度(低粘度SSG或低粒度CCS觀察到明顯的溶出增強,尤其是對于難溶性的藥物)。揭示了超級崩解劑的潛在生物制藥意義,發現藥物水溶性和介質特性之間的相互作用可能影響產品性能(由于崩解劑增加了水合能力,高溶性藥物在堿性介質中的藥物溶出速度比酸性介質快;難溶性藥物:與酸性介質相比,堿性介質中藥物溶出速度慢,這可能是由于片劑表面頂部存在高度溶脹的崩解劑結構)。SSG粘度、CCS粒度、藥物水溶性以及超級崩解劑差異被認為是影響藥物性能的關鍵生物制藥因素。結論是:崩解劑的差異對口服固體制劑的性能具有挑戰性,并且當在口服固體制劑開發中需要改變崩解劑類型/型號時,這是需要慎重考慮的因素。
九、參考文獻
略






