

時(shí)間:

翻譯:工業(yè)藥劑發(fā)燒友Summer
審核:華溶應(yīng)用中心
長(zhǎng)效注射混懸劑難溶性藥物晶體在體內(nèi)緩慢溶解,可將藥物體內(nèi)釋放時(shí)間延長(zhǎng)到數(shù)周至數(shù)月。迄今為止,F(xiàn)DA已經(jīng)批準(zhǔn)了大約10種長(zhǎng)效注射混懸劑,但大多數(shù)都沒(méi)有等效的仿制藥,很可能是由于復(fù)雜的處方工藝以及建立體外-體內(nèi)相關(guān)性(IVIVC)方面的困難。基于動(dòng)物模型的A級(jí)IVIVC已被證明可用于具有多相釋放特征的復(fù)雜長(zhǎng)效微球。長(zhǎng)效注射混懸劑的釋放特性相對(duì)簡(jiǎn)單,只有一個(gè)藥物溶出階段,因此可建立IVIVC。為了建立長(zhǎng)效注射混懸劑的體內(nèi)外相關(guān)性,以長(zhǎng)效注射醋酸甲羥孕酮(Depo-SubQ Provera 104)為上市參比制劑,制備了四種活性成分相同但處方和工藝(藥物粒度和輔料來(lái)源)不同的醋酸甲羥孕酮長(zhǎng)效注射混懸劑。然后使用兩種基于USP裝置二(帶有浸沒(méi)池)和USP裝置四(帶有半固體適配器)改進(jìn)的體外釋放測(cè)試方法,通過(guò)兔模型研究體內(nèi)釋放,并使用USP裝置四法獲得了體外釋放曲線,成功建立了A級(jí)IVIVC。
長(zhǎng)效注射混懸劑難溶性藥物晶體在體內(nèi)緩慢溶解,可將藥物體內(nèi)釋放時(shí)間延長(zhǎng)到數(shù)周至數(shù)月。迄今為止,F(xiàn)DA已經(jīng)批準(zhǔn)了大約10種長(zhǎng)效注射混懸劑,但大多數(shù)都沒(méi)有等效的仿制藥,很可能是由于復(fù)雜的處方工藝以及建立體外-體內(nèi)相關(guān)性(IVIVC)方面的困難。基于動(dòng)物模型的A級(jí)IVIVC已被證明可用于具有多相釋放特征的復(fù)雜長(zhǎng)效微球。長(zhǎng)效注射混懸劑的釋放特性相對(duì)簡(jiǎn)單,只有一個(gè)藥物溶出階段,因此可建立IVIVC。為了建立長(zhǎng)效注射混懸劑的體內(nèi)外相關(guān)性,以長(zhǎng)效注射醋酸甲羥孕酮(Depo-SubQ Provera 104)為上市參比制劑,制備了四種活性成分相同但處方和工藝(藥物粒度和輔料來(lái)源)不同的醋酸甲羥孕酮長(zhǎng)效注射混懸劑。然后使用兩種基于USP裝置二(帶有浸沒(méi)池)和USP裝置四(帶有半固體適配器)改進(jìn)的體外釋放測(cè)試方法,通過(guò)兔模型研究體內(nèi)釋放,并使用USP裝置四法獲得了體外釋放曲線,成功建立了A級(jí)IVIVC。
IVIVC是一種預(yù)測(cè)數(shù)學(xué)模型,用來(lái)研究不同劑型藥物的體外溶出與體內(nèi)釋放之間的關(guān)系。成功的IVIVC可用于制定釋放/溶出測(cè)試規(guī)范,當(dāng)建立了人體內(nèi)的A級(jí)相關(guān)性時(shí),可代替體內(nèi)BE研究。在藥物初次批準(zhǔn)過(guò)程中以及在獲批后出現(xiàn)微小變更(例如生產(chǎn)場(chǎng)所變更)時(shí),IVIVC可以減輕BE研究的臨床負(fù)擔(dān)。迄今為止,關(guān)于IVIVC的建立、評(píng)估和應(yīng)用,僅有一項(xiàng)針對(duì)緩釋口服劑型的FDA指南。因此在過(guò)去20余年里,科學(xué)家們不得不依賴(lài)這一指南來(lái)開(kāi)發(fā)非口服制劑。一項(xiàng)針對(duì)IVIVC研究的行業(yè)調(diào)研顯示,半數(shù)受訪者很少或從未建立過(guò)非口服劑型的IVIVC模型。FDA已批準(zhǔn)的長(zhǎng)效注射混懸劑產(chǎn)品中,只有Invega Sustenna提供了針對(duì)不同粒徑的臨床A級(jí)IVIVC 。近年來(lái),由于人體研究相對(duì)困難并且昂貴,非臨床動(dòng)物模型已被用于非口服劑型IVIVC的研究,可能為人類(lèi)IVIVC的建立鋪平道路,例如各種聚乳酸-甘氨酸微球制劑如地塞米松、利培酮、納曲酮、醋酸亮丙瑞林,已經(jīng)通過(guò)兔模型成功建立了A級(jí)(點(diǎn)對(duì)點(diǎn))IVIVC。
在IVIVC模型研究中,根據(jù)所選制劑的藥代動(dòng)力學(xué)特征,動(dòng)物模型或人體受試者的體內(nèi)吸收(血藥濃度-時(shí)間變化)呈現(xiàn)出恒定的規(guī)律。但當(dāng)使用不同的體外釋放/溶出方法(設(shè)備、釋放介質(zhì)、溫度、攪拌速度和上樣方式)時(shí),所選制劑的體外釋放曲線可能會(huì)有所不同。因此,選擇合適的體外釋放/溶出方法來(lái)建立IVIVC至關(guān)重要。盡管FDA已經(jīng)推薦了一些長(zhǎng)效注射混懸劑的體外釋放檢測(cè)方法,但它們的持續(xù)時(shí)間較短(30分鐘至2天),可能不足以建立用于長(zhǎng)效注射混懸制劑的IVIVC(體內(nèi)有效持續(xù)時(shí)間為數(shù)周至數(shù)月)。本文開(kāi)發(fā)了一種能夠延長(zhǎng)注射混懸劑體外釋放時(shí)間的方法,有望用于IVIVC的研究。
在IVIVC模型研究中,根據(jù)所選制劑的藥代動(dòng)力學(xué)特征,動(dòng)物模型或人體受試者的體內(nèi)吸收(血藥濃度-時(shí)間變化)呈現(xiàn)出恒定的規(guī)律。但當(dāng)使用不同的體外釋放/溶出方法(設(shè)備、釋放介質(zhì)、溫度、攪拌速度和上樣方式)時(shí),所選制劑的體外釋放曲線可能會(huì)有所不同。因此,選擇合適的體外釋放/溶出方法來(lái)建立IVIVC至關(guān)重要。盡管FDA已經(jīng)推薦了一些長(zhǎng)效注射混懸劑的體外釋放檢測(cè)方法,但它們的持續(xù)時(shí)間較短(30分鐘至2天),可能不足以建立用于長(zhǎng)效注射混懸制劑的IVIVC(體內(nèi)有效持續(xù)時(shí)間為數(shù)周至數(shù)月)。本文開(kāi)發(fā)了一種能夠延長(zhǎng)注射混懸劑體外釋放時(shí)間的方法,有望用于IVIVC的研究。
2.1 材料
DMSO (USP級(jí))購(gòu)自Sigma-Aldrich(St.Louis,MO,USA)。戊烷,磷酸氫二鉀,含0.1%甲酸的乙腈(v/v),LC/MS級(jí)Optima?和含0.1%甲酸(v/v)的水, LC/MS級(jí)Optima?購(gòu)自Fisher Scientific(Hampton,NH,USA)。Depo SubQ Provera 104mg醋酸甲羥孕酮注射混懸液(104mg/0.65mL)購(gòu)自Pfizer Inc。醋酸甲羥孕酮(微粉化,USP級(jí))購(gòu)自Spectrum Chemical Manufacturing Corp(New Brunswick,NJ,USA)。甲羥孕酮17-乙酸酯-d3(MPA-d3)購(gòu)自TLC Pharmaceutical Standard ltd(Ontario,Canada)。MiniCollect 0.8ml肝素分離管購(gòu)自Greiner Bio-One North America Inc。(Monroe,North Carolina,USA)。除非另有說(shuō)明,否則所有材料均為分析級(jí)。
2.2 MPA混懸液的制備及體外釋放數(shù)據(jù)
首先用四種方法制備MPA混懸劑 :使用MPA原料藥制備的制劑1(F1);使用重結(jié)晶制備的制劑2(F2)(反溶劑法:丙酮與水之比為1:1,分別作為良溶劑和反溶劑);通過(guò)探針超聲處理制備的制劑3(F3);制劑4(F4)除了使用不同來(lái)源的PEG 3350(F1:來(lái)自Spectrum Chemicals;F4:來(lái)自BASF)外與F1相同。使用馬爾文粒度儀測(cè)定所有制劑的粒徑,記錄Dv10,Dv50和Dv90,所有實(shí)驗(yàn)重復(fù)三次,然后計(jì)算跨度值((Dv90-Dv10)/Dv50)來(lái)評(píng)估混懸劑的粒度分布,所有數(shù)據(jù)均以均數(shù)±標(biāo)準(zhǔn)差表示。
所有MPA混懸劑的體外釋放數(shù)據(jù)均來(lái)自之前的一份報(bào)告,其中使用了兩種裝置,分別為帶有浸沒(méi)池(4cm2接觸面積)的USP裝置二(推薦使用華溶儀器DS-1206AT全自動(dòng)取樣溶出系統(tǒng))和帶有半固體適配器(1mm深度)的USP裝置四 (圖1)(推薦使用華溶儀器DS-7CP流池法溶出系統(tǒng))。
2.3 MPA混懸劑的體內(nèi)釋放研究
2.3.1 MPA混懸劑的動(dòng)物研究
以兔為模型研究了MPA混懸劑和商業(yè)產(chǎn)品Depo SubQ Provera 104在體內(nèi)的釋放情況。將體重約4kg的雌性新西蘭白兔隨機(jī)分配到每個(gè)制劑組(n=6)。將每種制劑以26mg/kg的劑量皮下注射(0.65mL)。為了獲得藥物PK參數(shù),MPA藥物溶液(在DMSO中)也以4 mg/kg的劑量進(jìn)行靜脈注射(n=6)。以預(yù)定的間隔從耳緣靜脈收集血液樣品并置于肝素分離管中。樣品在5000rpm的速度下離心5分鐘得到血漿,然后在-80℃環(huán)境下儲(chǔ)存直到分析。本研究動(dòng)物模型的選擇是基于以下幾點(diǎn)原因:(1)PK研究的持續(xù)時(shí)間長(zhǎng)達(dá)3個(gè)月,為確保有足夠的血容量用于長(zhǎng)時(shí)間的連續(xù)采樣,認(rèn)為較大的動(dòng)物(如兔子)比較合適;(2)MPA用于避孕,因此選擇雌性動(dòng)物。為保證動(dòng)物試驗(yàn)的安全性,本研究所采用的給藥劑量均按相關(guān)文獻(xiàn)規(guī)定,并確保給藥后的血藥濃度能被檢測(cè)出來(lái),本研究按照康涅狄格大學(xué)機(jī)構(gòu)動(dòng)物護(hù)理和使用委員會(huì)(IACUC)審查和批準(zhǔn)的方案進(jìn)行。
2.3.2 樣品制備
超高效液相色譜(UPLC)
采用Vanquish UPLC系統(tǒng)(Thermo Fisher Scientific, Waltham, MA, USA)進(jìn)行樣品分離。并使用Kinetex EVO C18 (50 mm*2.1 mm, 2.6μm)和帶有SecurityGuard?超濾筒的預(yù)柱(Phenomenex Inc, Torrance, CA, USA),將柱溫箱溫度設(shè)置為30℃。流動(dòng)相由兩種溶劑混合物組成,A為含0.1% v/v甲酸的水;B為含0.1% v/v甲酸的乙腈; LC/MS級(jí)Optima),用8分鐘梯度法(表1)以0.3 mL/min的流速洗脫系統(tǒng),進(jìn)樣體積為20μL。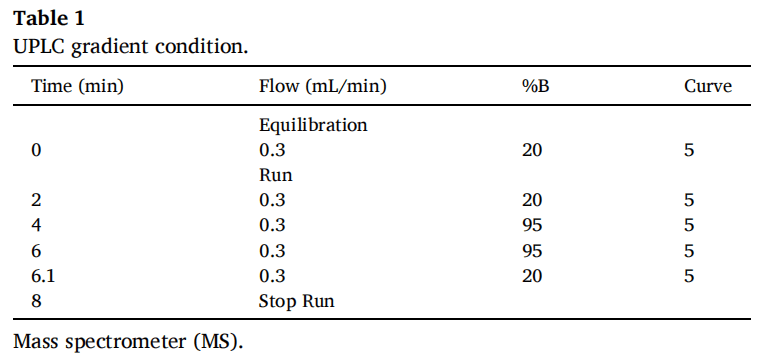
在全掃描模式下使用配備OptaMax?NG電噴霧(H-ESI)離子源(Thermo Fisher Scientific, Waltham, MA, USA)的Thermo Scientific?高分辨率Orbitrap Exploris?480質(zhì)譜儀 (質(zhì)量范圍:2500 - 425.00m/z,分辨率:120,000)。對(duì)MS參數(shù)進(jìn)行優(yōu)化來(lái)獲得目標(biāo)藥物分子的最佳強(qiáng)度。離子源參數(shù)設(shè)置如下:噴霧電壓為3700V的正離子模式;護(hù)套氣體流量為70Arb;輔助氣體流量為23Arb;掃掠氣體流量為5Arb;離子轉(zhuǎn)移管溫度為400°C;汽化器溫度為525°C。方法總時(shí)間為8min。藥物MPA和內(nèi)標(biāo)(MPA-d3)的親本離子質(zhì)量分別為387.2525m/z和390.2700m/z。通過(guò)Thermo Scientific Xcalibur?軟件(4.4版本)進(jìn)行數(shù)據(jù)采集和分析。
MPA和內(nèi)標(biāo)的保留時(shí)間均為4.81min。除最低濃度(0.5ng/mL)的回收率約為70%外,其他濃度MPA的回收率均在90%-101%范圍內(nèi)。校準(zhǔn)曲線建立在0.5-50 ng/mL和50-1000ng/mL范圍內(nèi),線性良好(R2>0.9995),準(zhǔn)確度小于15%,精密度小于10%。
2.4 統(tǒng)計(jì)分析
使用OriginPro 2017軟件(OriginLab Corporation)進(jìn)行線性回歸和擬合。數(shù)據(jù)以平均值±標(biāo)準(zhǔn)偏差(SD)表示。
2.5 IVIVC建立
為了研究用于MPA混懸劑的IVIVC,使用Phoenix WinNonlin軟件和IVIVC工具包(版本6.4,Certara Inc.,NJ,USA)進(jìn)行數(shù)據(jù)分析。目前的研究集中在A級(jí)IVIVC(體外溶出度與體內(nèi)釋放速率之間的點(diǎn)對(duì)點(diǎn)關(guān)系),因此它是最具參考性的,以下是數(shù)據(jù)分析的三個(gè)主要步驟。
步驟1:體外數(shù)據(jù)輸入
步驟2:體內(nèi)釋放數(shù)據(jù)輸入
在軟件中通過(guò)數(shù)據(jù)反卷積處理獲得所選MPA混懸劑的體內(nèi)釋放曲線。以靜脈注射劑的藥代動(dòng)力學(xué)特征為參考。設(shè)定單位脈沖響應(yīng)(UIR)指數(shù)的最大值為2,采用Akaike模型進(jìn)行數(shù)據(jù)反卷積。
步驟3:預(yù)測(cè)和驗(yàn)證相關(guān)性
通過(guò)軟件計(jì)算得到吸收比例因子(scaling factor for absorption)、時(shí)間比例因子(scaling factor for time)、時(shí)移因子(time shifting factor),用不同的縮放和/或移位因子建立體外和體內(nèi)釋放數(shù)據(jù)之間的相關(guān)性,在此基礎(chǔ)上對(duì)所建立的IVIVC進(jìn)行驗(yàn)證,并對(duì)AUC和Cmax的預(yù)測(cè)誤差百分比(%PE)進(jìn)行分析。根據(jù)FDA口服緩釋劑型IVIVC研究指南 ,IVIVC % PE的合格標(biāo)準(zhǔn)是:(1)對(duì)于體內(nèi)預(yù)測(cè)性,Cmax和AUC的平均% PE≤10%。此外,每種制劑的%PE不應(yīng)超過(guò)15%;(2)如果體內(nèi)預(yù)測(cè)性不確定,則應(yīng)通過(guò)體外預(yù)測(cè)性來(lái)最終確定IVIVC是否能夠替代BE;(3)對(duì)于體外預(yù)測(cè)性,Cmax和AUC的%PE應(yīng)≤10%。
3.1 粒徑與粒徑分布
表2列出了MPA混懸劑(F1、F2、F3和F4)及RLD的粒徑和粒徑分布。排列順序如下:F3
3.2 所有制劑的體內(nèi)藥物釋放
以家兔為實(shí)驗(yàn)對(duì)象,對(duì)MPA混懸劑和RLD進(jìn)行體內(nèi)藥物釋放研究。為了獲得PK曲線,所有制劑均經(jīng)皮下注射 (圖2B)。還進(jìn)行靜脈注射藥物溶液(圖2A)用于反卷積處理獲得PK參數(shù)。為了獲得PK曲線的清晰視圖,圖3A顯示了平均值曲線。使用Phoenix WinNonlin對(duì)所有制劑的體內(nèi)釋放曲線進(jìn)行反卷積。反卷積建立在非區(qū)室分析(NCA)的基礎(chǔ)上,詳細(xì)的方程推導(dǎo)可以在(Certara.com)中找到。為便于比較,還展示了先前報(bào)道的測(cè)試制劑的平均體外釋放曲線(圖3CD),粒徑是分散體系(混懸劑、納米顆粒、脂質(zhì)體)中的重要屬性,本研究重點(diǎn)考察了藥物粒徑對(duì)長(zhǎng)效注射混懸劑在體外和體內(nèi)釋放特性的影響。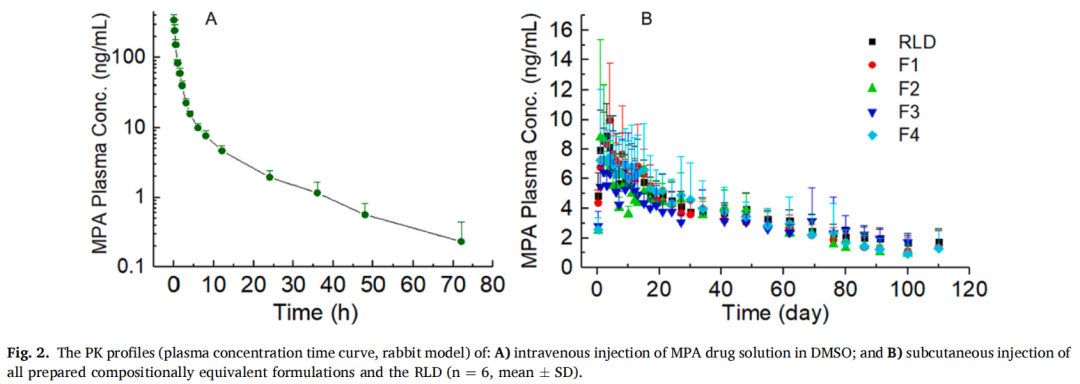
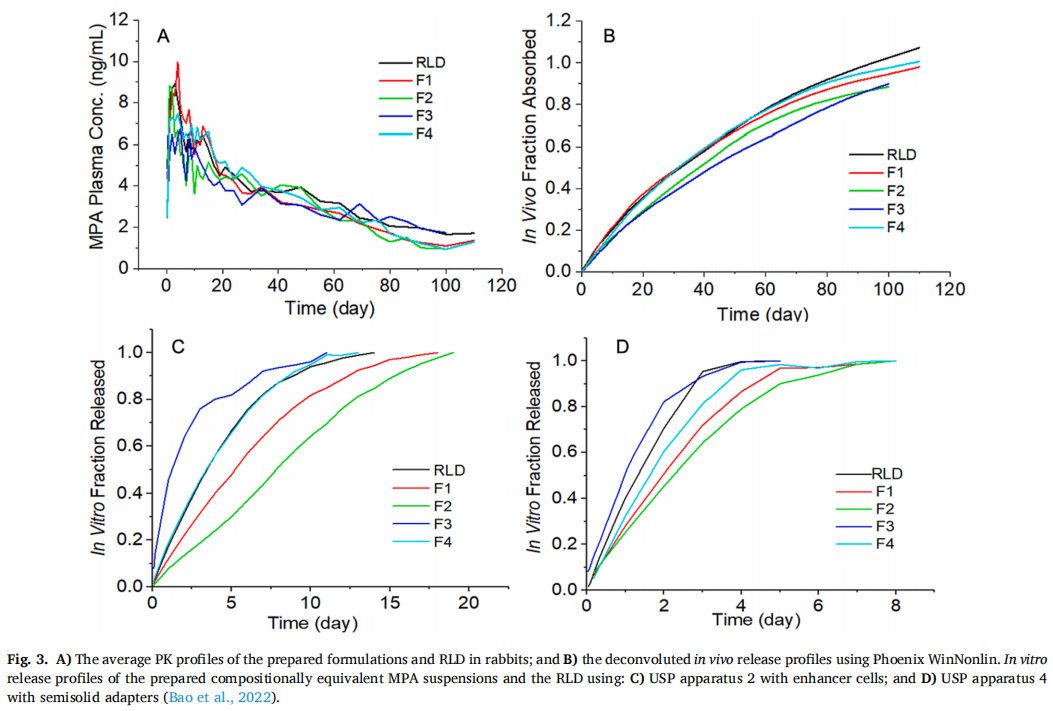
正如預(yù)期的那樣,藥物的粒徑顯著影響了體外和體內(nèi)的釋放曲線。粒徑越大,在體內(nèi)外的藥物釋放速率越慢。F1,F(xiàn)2,F(xiàn)4和RLD的體內(nèi)與體外釋放顯示出相同的次序(圖3CD)。但是F3并沒(méi)有遵循相同的規(guī)律,它的粒徑最小,預(yù)計(jì)在體內(nèi)釋放最快,相反,F(xiàn)3在體內(nèi)藥物釋放最緩慢。進(jìn)一步的研究表明,F(xiàn)3在體外并不穩(wěn)定,室溫下隨著時(shí)間的推移容易發(fā)生聚集,其粒徑(Dv50)在5天的時(shí)間內(nèi)一直增加到20 μm左右(圖4A)。此外,F(xiàn)3的跨度值也從2.61下降到1.5以下 (圖4B),表明F3的粒徑分布隨著時(shí)間的推移而變窄。因此,F(xiàn)3顆粒在注射部位的聚集是造成體內(nèi)釋放緩慢的重要原因之一。此外,也有可能F3在體內(nèi)快速溶解,然后在注射部位沉淀或再結(jié)晶,形成了更大粒徑的顆粒,從而減緩藥物釋放。由于建立IVIVC的最基本原則是體外和體內(nèi)的結(jié)果應(yīng)一致,因此F3并未包括在體內(nèi)外相關(guān)性的研究中。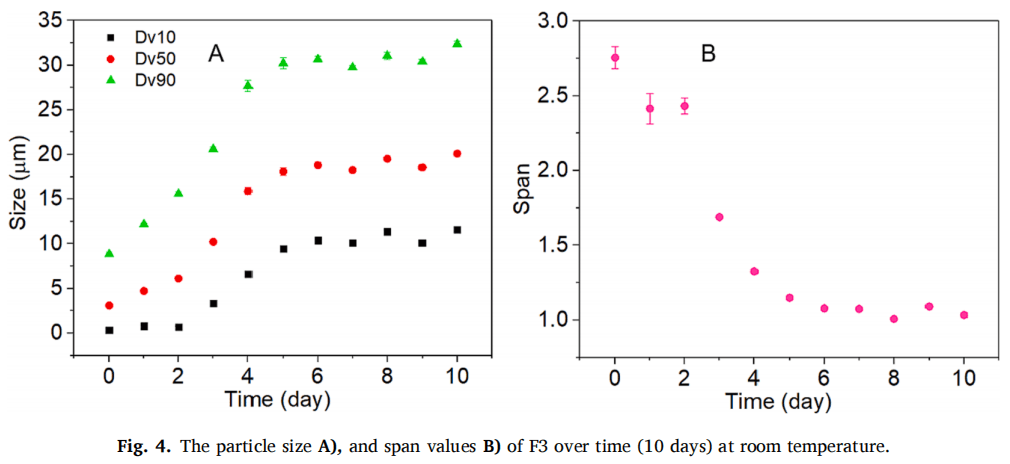
通過(guò)調(diào)整藥物粒徑(F1、F2和F3)和改變PEG 3350的賦形劑來(lái)源(F1和F4),制備了具有不同釋放速率的MPA混懸劑,雖然使用完全相同的懸浮介質(zhì),但藥物顆粒不一定具有物理穩(wěn)定性,以探針超聲法制備的F3為例,在常溫常壓條件下粒徑隨時(shí)間增長(zhǎng),初始粒徑(Dv50)約為3μm,到第5天粒徑約為20μm。在體外釋放試驗(yàn)中并未觀察到粒徑的變化,這很可能是因?yàn)樵诼┎蹢l件下釋放介質(zhì)具有稀釋效應(yīng),但在兔體內(nèi)釋放實(shí)驗(yàn)中觀察到藥物顆粒的聚集效應(yīng)。這些結(jié)果說(shuō)明了不同的工藝條件可以影響粒子的穩(wěn)定性,所以在不同工藝條件下制備混懸劑是具有挑戰(zhàn)性的,表明了精確控制長(zhǎng)效注射混懸劑粒徑的重要性。
3.3 MPA混懸劑的IVIVC研究
根據(jù)之前關(guān)于MPA混懸劑釋放測(cè)試方法開(kāi)發(fā)的報(bào)道,使用帶浸沒(méi)池的USP裝置二(圖3C)和帶半固體適配器的USP裝置四(圖3D)顯示出良好的釋放特性,可以嘗試建立A級(jí)IVIVC。使用兩種裝置可獲得較長(zhǎng)時(shí)間釋放曲線 (至少一周),具有可接受的再現(xiàn)性和區(qū)分不同粒徑MPA混懸劑的能力。在A級(jí)IVIVC的研究中,理想情況是體內(nèi)釋放部分與體外溶出部分之間有1:1的關(guān)系。如果體外釋放持續(xù)時(shí)間接近制劑的體內(nèi)療效持續(xù)時(shí)間,則有可能建立無(wú)時(shí)間縮放和轉(zhuǎn)換或最小時(shí)間縮放和轉(zhuǎn)換的IVIVC。與目前FDA推薦的用于其他長(zhǎng)效注射混懸劑產(chǎn)品的方法(30分鐘至2天)相比,這兩種方法體外釋放持續(xù)時(shí)間長(zhǎng)得多(USP裝置二為2周,USP裝置四為1周),但仍比體內(nèi)療效持續(xù)時(shí)間(3個(gè)月)短得多(圖3CD)。
使用WinNonlin IVIVC工具包進(jìn)行MPA混懸劑的IVIVC研究,將體外數(shù)據(jù)與所選制劑的體內(nèi)釋放數(shù)據(jù)進(jìn)行比較。
3.3.1 使用USP裝置二(帶浸沒(méi)池)獲得的體外釋放數(shù)據(jù)
首先,從帶浸沒(méi)池的USP裝置二中獲得了四種制劑F1, F2, F4和RLD的體外數(shù)據(jù)。F2、F4和RLD作為體內(nèi)制劑,F(xiàn)1作為體外制劑,采用雙Weibull模型擬合效果最佳。通過(guò)縮放和移位的方法,吸收分?jǐn)?shù)和釋放分?jǐn)?shù)之間的IVIVC線性回歸擬合系數(shù)(R2值)為0.9377(圖5A)。使用建立的IVIVC將預(yù)測(cè)與觀察到F1在體內(nèi)的釋放情況進(jìn)行比較(圖5B)。結(jié)果表明兩條曲線在一些時(shí)間間隔內(nèi)重疊,但在其他時(shí)段具有明顯的區(qū)別。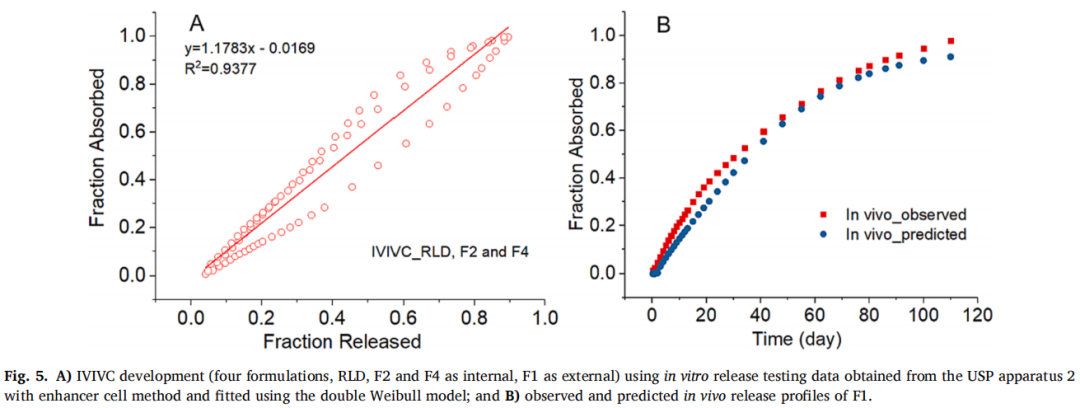
吸收比例因子(scaling factor for absorption)和時(shí)間比例因子(scaling factor for time)分別為0.91和0.17,時(shí)移因子(time shifting factor)為0.31。IVIVC驗(yàn)證總結(jié)如表3所示。對(duì)于體內(nèi)預(yù)測(cè)性,AUC的%PE符合標(biāo)準(zhǔn)(個(gè)體%PE<15%,平均%PE<10%),但Cmax的%PE>10%。因此使用USP裝置二(帶浸沒(méi)池)獲得的體外釋放數(shù)據(jù)并不能建立A級(jí)IVIVC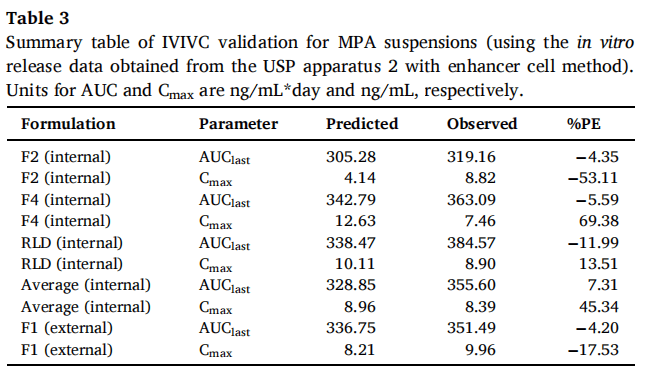
3.3.2 使用USP裝置四(帶半固態(tài)適配器)獲得的體外釋放數(shù)據(jù)
使用USP裝置四方法獲得體外釋放數(shù)據(jù),選擇三種或四種制劑用于IVIVC的研究。
3.3.2.1 三種制劑(F1、F4和RLD)
F1和RLD作為體內(nèi)制劑,F(xiàn)4作為體外制劑,Hill和Weibull模型均較好地?cái)M合了體外釋放曲線,僅使用縮放方法就能建立和驗(yàn)證IVIVC。Hill和Weibull模型的IVIVC線性回歸擬合系數(shù)(R2值)分別為0.9586和0.9599(圖6A和6C)。通過(guò)建立的IVIVC對(duì)F4制劑的體內(nèi)釋放曲線進(jìn)行預(yù)測(cè)和觀察,除了曲線末端的幾個(gè)時(shí)間點(diǎn)外均顯示出良好的一致性 (圖6B和6D)。采用Hill和Weibull模型,吸收比例因子(scaling factor for absorption)分別為0.91和0.93,時(shí)間比例因子(scaling factor for time)均為0.06。表4通過(guò)體外釋放數(shù)據(jù)的Hill和Weibull模型對(duì)IVIVC驗(yàn)證進(jìn)行了總結(jié)。對(duì)于不同的模型,IVIVC的預(yù)測(cè)性略有差異,比如使用Hill和Weibull模型,由于F1制劑Cmax的%PE絕對(duì)值較高,體內(nèi)預(yù)測(cè)性是不準(zhǔn)確的。但對(duì)于AUC和Cmax,體外預(yù)測(cè)性的%PE均在10%以?xún)?nèi)(Hill模型:AUC和Cmax的%PE分別為-9.81%和7.67%;Weibull模型:AUC和Cmax的%PE分別為- 8.61%和-9.76%)。根據(jù)FDA關(guān)于IVIVC緩釋片口服劑型申請(qǐng)指南,當(dāng)體內(nèi)預(yù)測(cè)性不準(zhǔn)確時(shí),若體外預(yù)測(cè)性的AUC和Cmax的%PE絕對(duì)值均小于10%,可采用體外預(yù)測(cè)性進(jìn)行IVIVC的評(píng)價(jià)。因此,使用F1、F4和RLD三種制劑成功建立了A級(jí)IVIVC,并使用USP裝置四獲得了體外釋放數(shù)據(jù)。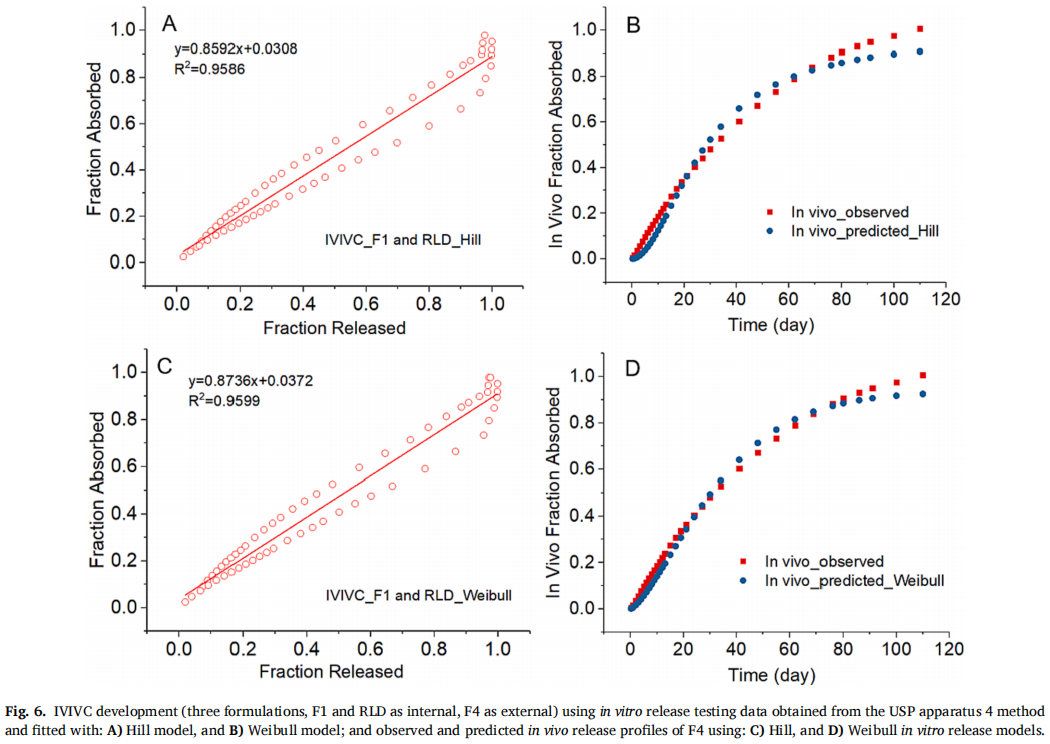
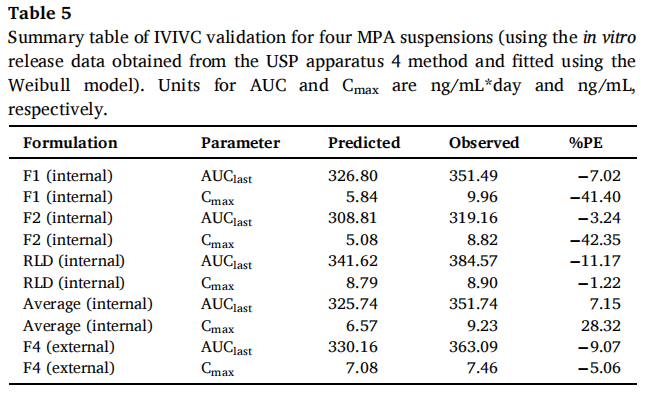
3.3.2.2 四種制劑(F1、F2、F4和RLD)
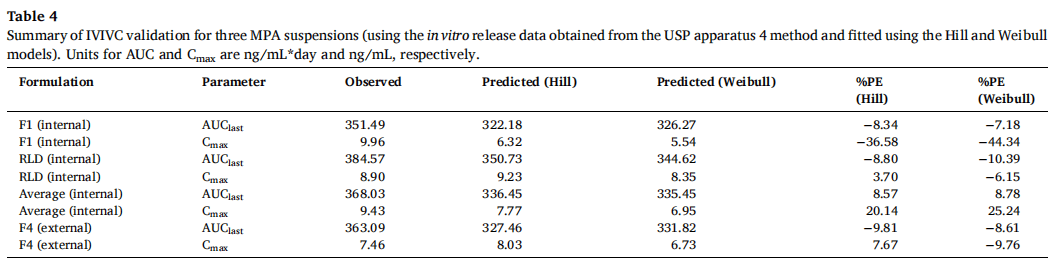
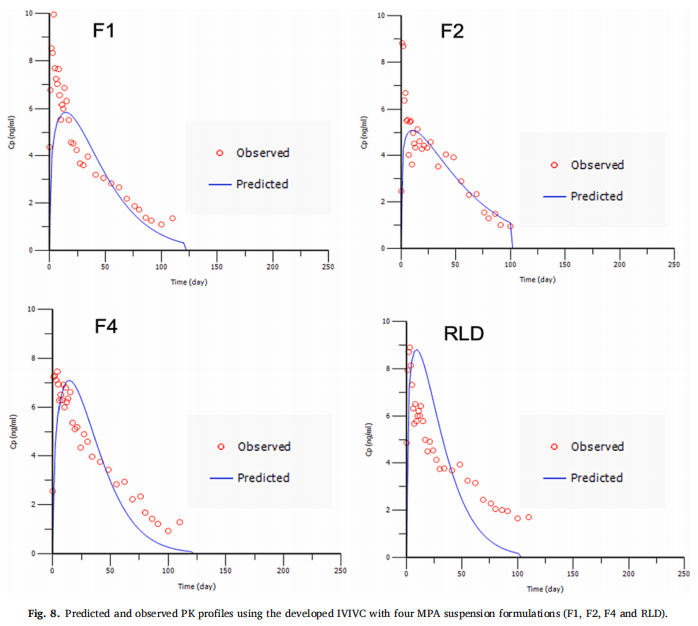
F1、F2和RLD作為體內(nèi)制劑,F(xiàn)4作為體外制劑。使用Hill和Weibull模型均可獲得擬合度高的體外釋放曲線。但使用Hill模型擬合的體外數(shù)據(jù)獲得IVIVC體外預(yù)測(cè)性的%PE值略超出標(biāo)準(zhǔn)(AUC的%PE值為-9.49%,Cmax的%PE值為12.26%)。因此,在IVIVC建立過(guò)程中選擇Weibull模型進(jìn)行體外釋放數(shù)據(jù)進(jìn)行建模,建立IVIVC并使用縮放方法進(jìn)行驗(yàn)證。使用F1、F2和RLD,吸收和釋放分?jǐn)?shù)之間的線性回歸擬合系數(shù)(R2值)為0.9664(圖7A)。觀察和由IVIVC預(yù)測(cè)的體內(nèi)曲線在多數(shù)時(shí)間段有重疊(圖7B)。吸收和時(shí)間比例因子分別為0.92和0.06。IVIVC驗(yàn)證總結(jié)如表5所示。與使用三種制劑建立的IVIVC類(lèi)似,F(xiàn)1和F2 %PE絕對(duì)值較高,體內(nèi)預(yù)測(cè)性不確定。但AUC(-9.07%)和Cmax(-5.06%)的%PE均在體外預(yù)測(cè)性的誤差范圍內(nèi)(10%)。因此,使用四種MPA混懸劑和從USP裝置四獲得的體外釋放數(shù)據(jù)成功建立了A級(jí)IVIVC,并且比較了四種制劑預(yù)測(cè)和觀察到的PK曲線(圖8)。由于Cmax的 %PE絕對(duì)值高,低估了F1和F2的Cmax預(yù)測(cè)值。相反,F(xiàn)4和RLD的Cmax預(yù)測(cè)值與觀察到的Cmax顯示出良好的一致性。由于目前關(guān)于長(zhǎng)效注射混懸劑的IVIVC研究文獻(xiàn)報(bào)道很少,這類(lèi)制劑的Cmax中%PE較高的原因尚不清楚,預(yù)測(cè)誤差較大可能是因?yàn)椋海?)建模所用的數(shù)據(jù)為平均值,Cmax值可能發(fā)生偏移;(2)體外與體內(nèi)釋放條件有很大區(qū)別,而且要同時(shí)考慮到給藥過(guò)程中對(duì)制劑產(chǎn)生的全部生理及生物學(xué)效應(yīng)很難做到;(3)隨著時(shí)間的推移,制劑可能在注射部位發(fā)生變化,例如沉淀、再結(jié)晶和聚集。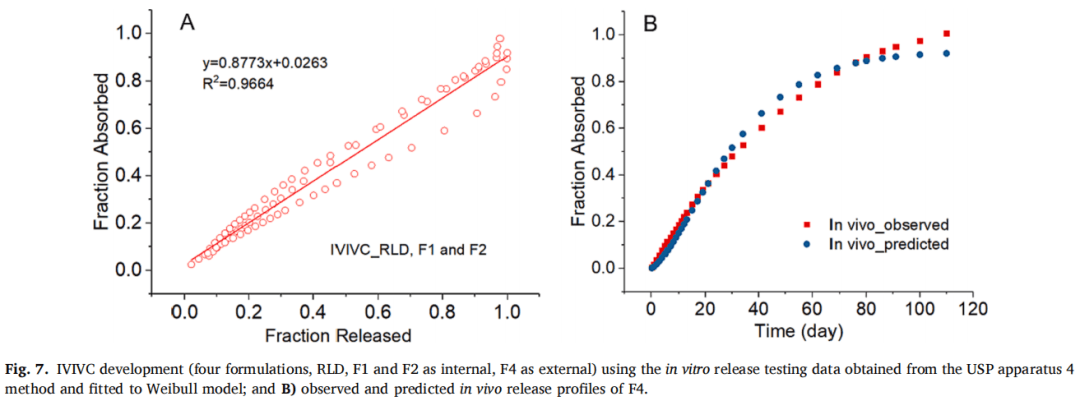
3.4 IVIVC研究的考慮因素
3.4.1 體外釋放模型
本研究使用三種不同的模型(Hill、Weibull和雙Weibull)進(jìn)行體外釋放曲線的數(shù)據(jù)分析。USP裝置二獲得的釋放曲線在使用雙Weibull模型時(shí)擬合最佳,而USP裝置四獲得的釋放曲線在使用Hill和Weibull模型時(shí)擬合最佳。在通過(guò)三種制劑(F1、F4和RLD)研究IVIVC的過(guò)程中,使用USP裝置四和Hill或Weibull模型成功建立了A級(jí)IVIVC。但通過(guò)四種制劑(F1、F2、F4和RLD)研究IVIVC時(shí),只有使用Weibull模型擬合體外數(shù)據(jù)才能建立A級(jí)IVIVC,因此選擇合適的模型對(duì)IVIVC研究至關(guān)重要。
3.4.2 體內(nèi)釋放數(shù)據(jù)和IVIVC研究
FDA指南并未規(guī)定建立IVIVC所需要的方法,只要所用模型在不同的IVIVC類(lèi)別下具有可接受的%PE即可。通過(guò)IVIVC開(kāi)發(fā)的長(zhǎng)效注射混懸劑均能滿(mǎn)足所有制劑的AUC標(biāo)準(zhǔn)(個(gè)體%PE<15%)。但是一些體內(nèi)和體外制劑Cmax的%PE絕對(duì)值高,導(dǎo)致通過(guò)USP裝置二無(wú)法建立A級(jí)IVIVC。相比之下,盡管USP裝置四的體內(nèi)預(yù)測(cè)性不準(zhǔn)確,但仍成功建立了A級(jí)IVIVC。
這是關(guān)于長(zhǎng)效注射混懸劑臨床前(兔模型)IVIVC研究的第一份報(bào)告。通過(guò)USP裝置四使用三種或四種制劑獲得了體外釋放數(shù)據(jù),成功建立了A級(jí)IVIVC。由于制劑的Cmax的預(yù)測(cè)誤差值較高,不能使用USP裝置二獲得的體外數(shù)據(jù)來(lái)建立IVIVC,因此在長(zhǎng)效注射混懸劑的體外釋放方法開(kāi)發(fā)過(guò)程中,出于產(chǎn)品質(zhì)量控制和IVIVC研究的目的,可以?xún)?yōu)先考慮USP裝置四方法。藥物粒徑顯著影響體外和體內(nèi)釋放過(guò)程,被認(rèn)為是長(zhǎng)效注射混懸劑開(kāi)發(fā)中的一個(gè)關(guān)鍵質(zhì)量屬性。體外釋放模型的選擇(Hill、Weibull和雙 Weibull等)可能會(huì)改變IVIVC建立的最終結(jié)果,因此在數(shù)據(jù)分析中必須仔細(xì)考慮。所建立的IVIVIC將為長(zhǎng)效注射混懸劑的處方篩選和優(yōu)化,以及體外釋放方法研究提供科學(xué)依據(jù)。
略






