

時間:

胃腸道(GI)是最受歡迎使用的給藥途徑之一,因為與其他途徑相比,胃腸道可以提高患者的依從性并降低成本。然而,在開發更復雜的劑型時,如結合兩種或多種藥物的劑型,其復雜的性質帶來了巨大的挑戰。固定劑量復方制劑(FDC)是兩種或多種單一活性成分組合在一個單一劑型中。這種處方組成代表了一種新的處方,與單獨的每種單一產品相比,它是安全有效的。通過復雜途徑給藥的復雜藥物產品,需要對配方開發進行深入的考慮。此外,在證明此類仿制藥的生物等效性(BE)時,從監管角度來看,這是一個挑戰。本報告向讀者簡要介紹了2018年11月3日和4日在華盛頓舉行的藥物科學家年度協會(AAPS)會議上進行的為期兩天的短期課程。這篇手稿將全面了解胃腸道生理學對藥物吸收最有影響的方面,以及目前的技術,以幫助理解口服藥物產品在以胃腸道為代表的復雜環境中的過程。通過對FDC產品開發和監管問題的案例研究,本文將為讀者提供一個探索開發FDC產品的常規方法。
從解剖學的角度來看,胃分為胃底、胃體和胃竇,但就運動功能而言,可以分為兩部分:由胃底和胃體近端組成的近端胃和由胃體遠端和胃竇組成的遠端胃。近端胃的運動特征是平滑肌(張力)保持收縮狀態,而遠端胃產生階段性收縮。在消化間期,胃近端肌張力高,而遠端胃處于一種周期性運動模式,稱為遷移運動復合體(MMC)。這種復合體涉及胃和大部分小腸(但不包括遠端小腸),分為三個階段:第一階段,無收縮的靜止期;第二階段,隨機收縮;第三階段,突然出現的重復性收縮,也會突然結束;第三階段可以在胃或小腸近端開始,然后向回腸遠端遷移。胃pH在MMC期間波動,胃竇pH在第三階段收縮開始前最低(酸性更強),在第一階段開始時較高。這種pH值的變化是由于MMC的第三階段以及十二指腸的無膽汁碳酸氫鹽回流導致的酸和胃蛋白酶分泌增加。腸道和胰腺分泌物(如水、碳酸氫鹽和胰酶)在小腸第三期收縮期間增加。一旦食物被攝入,近端胃就會放松以容納食物,然后近端胃就會緊張性收縮,將食物推向更遠的地方。遠端胃會通過有力而有規律的收縮來混合和研磨食物。十二指腸在攝入食物后幾乎直接暴露于營養物質中,這將激活多種十二指腸負反饋機制,例如通過迷走神經反射和激素信號介導的負反饋機制。這將通過抑制近端胃張力和收縮,刺激幽門閉合,從而延遲酸性、高滲或高熱量胃內容物進入十二指腸。食物的物理狀態一致性、脂肪含量和熱量負荷在調節胃的運動反應中起著相關的作用。低熱量的液體在由胃底張力和遠端胃的少量運動以指數方式產生的壓力梯度下排空。更黏稠的食物需要經胃竇研磨,直至粒度減小。胃減少顆粒所需的時間可以解釋排空開始前觀察到的所需時間。因此,胃排空分為兩個階段:緩慢排空期(負責固體物質的消化)和排空期即消化后的固體顆粒或液體可以很容易地從胃中排空的線性排空期。不可消化的固體通常在MMC的消化I第三階段從胃中排空。最近的一項研究證明了這些第三階段收縮對將布洛芬從胃排空到小腸產生影響。這些收縮在腸道吸收中pH起著關鍵作用,決定了Cmax和Tmax。最近進行了一項臨床排空研究,以研究禁食和進食狀態下一杯水的胃排空率。將標準劑量的酚紅添加到一杯水中,并由健康受試者攝入。喝了這杯水后,從胃、十二指腸和空腸抽出胃腸液。基于計算模型,作者發現,一杯水的胃排空非常迅速,尤其是在禁食狀態下。
閃爍掃描成像被認為是研究人類胃排空的金標準,通常由1小時、2小時和4小時的胃滯留百分比來定義。然而,使用單一的測量結果總結并不能獲得上述由飲食激活的復雜機制的報告。正如Parker及其同事所描述的,諾丁漢大學已經驗證并公布了基于液體餐的胃排空測試的正常值,以獲得對胃運動和感知功能的全面評估。該測試可以區分液體餐時的胃排空的早期和晚期,這可以反映胃的調節和胃竇的排空。近年來發展了兩種研究胃功能的技術。SmartPill?是一種可攝入的設備(26mm × 13mm),可測量腔內pH值、壓力和溫度。它將數據無線傳輸到一個可穿戴的外部記錄器,允許在家進行動態研究。腸道pH值的變化,以及排便后溫度的下降,可以精確測量區域和整個腸道的轉運時間。然而,需要注意的是,考慮到該裝置的尺寸并不能反映正常可消化食物的胃排空情況,而且確實發現胃排空時間比閃爍成像測量的時間更長。無論如何,該技術具有非侵入性以及結合整個腸道轉運過程pH值測量的優點。除了考察膠囊外,磁共振成像(MRI)最近也被應用于胃腸道功能的研究,與其他技術相比,這項技術具有一些主要優勢:它是非侵入性的,不會使受試者暴露于輻射中,并且不需要任何造影劑。這是一種可以同時測量胃、小腸和結腸的體積、物理化學特征以及轉運速率,并量化運動方式的獨特的技術。這項技術尚未在各研究中心建立標準化,目前還不允許評估直立狀態下的胃功能。總之,人類的胃看起來更復雜,并且在體內藥物行為以及腸道吸收中發揮主要作用。
除了胃,小腸在藥物吸收方面發揮關鍵作用。特別關注(i)腸液體積,(ii)腸液的特征和組成,以及(iii)腸壁對藥物化合物的滲透性。腸道內的殘液量相當少,而且不像一池水一樣從近端向遠端均勻分布。這些液體合理的分布在不同的流體庫中。正如Mudie及其同事所強調的那樣,健康受試者的儲庫數量和每個儲庫的實際體積差異非常大。這一發現是一項重要的研究,讓配方科學家意識到腸道不像一個“游泳池”,完全充滿了水。根據Mudie等人的觀察,使用流體動力學而不是靜態和大體積來預測口服藥物的體內特性更為準確。這是在泊沙康唑(一種弱堿性化合物)中觀察到的,通過使用模擬軟件中的動態流體和pH模型來預測其體內性能。盡管該模型對預測水溶性較差的化合物的體內性能有影響,但作者得出結論,該模型對預測溶解度較高的化合物的全身暴露可能沒有很大影響。此外,如前所述,在儲庫的數量和體積上存在巨大的差異。例如,一名受試者顯示只有2個儲庫,總容積為1.4 mL,而另一名受試者顯示有23個儲庫,總容積為160 mL。一項后續研究旨在揭示液體庫的出現與當前運動特性之間的潛在聯系。20世紀70年代,Vantrappen等人在上消化道第三階段收縮后不久觀察到碳酸氫鹽的分泌率較高。這樣,進入小腸的胃酸可以直接被碳酸氫鹽中和。這種所謂的促分泌物很可能是腸道內形成液體儲液庫的一個重要因素。除了獲得有關胃腸道中體積的知識外,這些流體的組成是另一個重要方面。在最近的一項研究中,從20名處于禁食和進食狀態的健康受試者身上抽取人類十二指腸液。通過攝入液體膳食(即400 mL的Essure Plus?,等于700卡路里)來模擬進食狀態。在獲取這些液體作為時間的函數后,分析液體的pH值和內源性成分(膽汁鹽、磷脂、膽固醇、酶活性和脂質消化產物)。這項研究的結果表明,這些成分的存在因人而異,盡管每個人的研究方案都是相同的。特別是對于離子化的化合物,腸道中的pH值對于藥物的溶解和隨后的吸收是極其重要的。Amidon教授(密歇根大學)的研究小組在禁食和進食狀態下口服布洛芬緩釋片(800 mg)后,從37名健康受試者身上吸出胃腸道液體。從胃腸道的不同部分吸出液體:胃、十二指腸和空腸。本研究顯示,pH值的高度波動,特別是在十二指腸,這是除運動外的一個重要內在因素,解釋了受試者之間和受試者內部布洛芬全身暴露的差異(圖1)。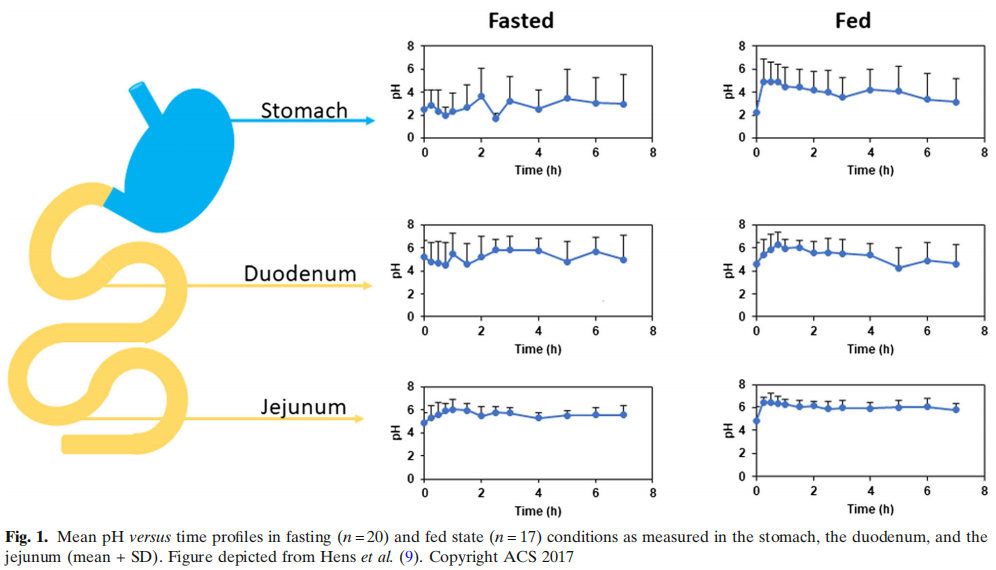
除了溶解度外,吸收一直是評估藥物性能的關鍵參數。文獻中描述了多種技術來評估藥物化合物的腸道通透性。文獻中描述了多種技術來評估藥物化合物的腸道滲透性。Loc-l-Gut?方法,即雙球囊灌注系統,是一種有趣的研究技術,用于探索藥物化合物在胃腸道不同區域的滲透性。胃腸道的特定區域將被兩個球體所膨脹,從而分離出基于性質的特定區域。隨后,將灌流藥物溶液,消失的藥物量是衡量藥物吸收量的指標。這項技術的應用已經揭示了氫化可的松在十二指腸、空腸和回腸的腸道滲透性。Dahlgren等人最近的一篇綜述匯編了在人類腸道所有部位進行的61項研究中對80種物質進行的273次單獨測量的歷史Peff數據。這一令人印象深刻的數據為研究人員提供了參考,以優化體外研究的方案,從而提高其內部吸收的預測。
關于結腸生理學,應用高分辨率測壓法(HRM)的最新發現表明,腸運動主要表現為非傳遞和逆行向后運動,并且這兩種活動在進食后不久都會增加。這些結腸運動模式具有延遲結腸內容物到達直腸的作用,并有利于橫結腸和升結腸的充盈,而橫結腸和升結腸通常是傳播性收縮開始的地方。傳播性收縮,包括帶有固體內容物結腸運動相關的高振幅傳播性收縮代表了結腸活動的一小部分,通常在飯后約1-2小時和醒來時更頻繁。其原因很可能與以下事實有關:在白天的這些時刻,夜間和消化間期積聚在遠端小腸的內容物的到達決定了升結腸和橫結腸的膨脹,從而觸發了傳播活動。非傳播性運動的普遍存在解釋了與小腸相比,正常結腸運輸時間較慢(約35小時)的事實。這使結腸能夠發揮其吸收和發酵的功能,并成為一個足夠大的儲存器官。HRM是研究結腸運動功能的一種有用技術,但具有侵入性,通常需要腸道特殊準備。這使得當需要在生理條件下研究結腸功能時,該技術的吸引力降低。最近,其他技術已被應用于結腸功能的研究。電磁膠囊是一種可攝入的涂有硅膠的圓柱形磁體(21毫米x 8毫米),用于繪制結腸內容物的實時運動圖。一個裝有4x4磁場傳感器檢測平板佩戴在患者腹部周圍,以監測藥物的運動。該方式將藥物運動映射在x、y和z軸上以及結腸施加的傾斜角度。該藥物可以評估腸道內內容物的運動方向(順時針和逆時針)、速度和距離,從而計算結腸傳輸時間。最近的研究也證明了結腸運動模式與HRM研究結果一致。此外,還引入了MRI,因為它能夠測量結腸游離水含量和結腸內容物的“流動性”。最近的動物研究表明,結腸能夠適應內容物的各種物理特征,并根據或多或少的液體內容物的存在產生不同的運動反應。這些生理變量很可能在藥物的溶解和/或吸收中發揮關鍵作用,這些藥物在人類胃腸道的結腸部位釋放。
胃腸道是一個復雜的器官,其環境隨著時間的變化而變化。然而,對每個環節的難點和機會進行深入的理解對于實現最佳藥物吸收和生物利用度(BA)是必要的(圖2)。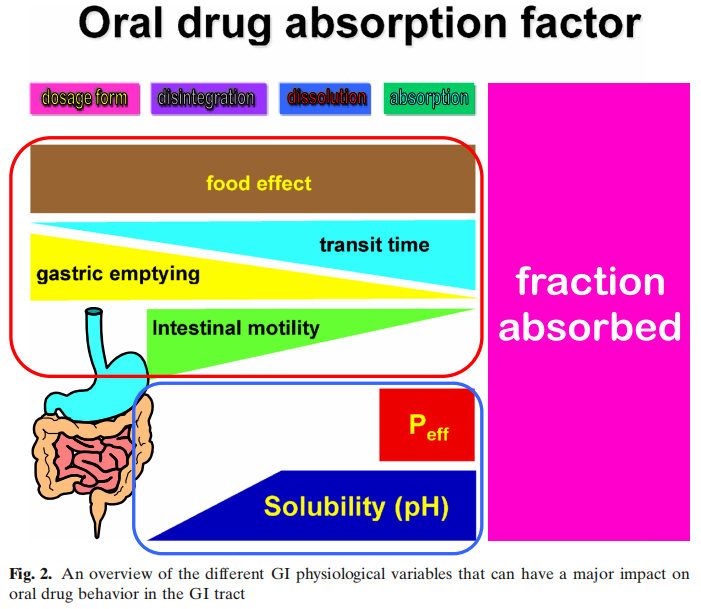
圖2顯示了影響吸收的多個因素,而沒有考慮代謝或藥物穩定性的影響。以藍色方框表示的生物制藥分類系統(BCS)側重于滲透性和溶解性。然而,藥物的溶解性和溶解度取決于紅框中總結的生理因素。眾所周知,運動效應和胃排空會對藥物的性能產生影響,但在藥物開發中很少考慮它們。相反,在過去的幾十年里,人們對食物效應、pH效應和膽汁鹽的增溶作用進行了深入的研究。然而,今天,對于一種通用的溶出介質仍然沒有達成共識,這種介質可以用于藥物開發和體外性能測試,以獲得這些結果。早期研究評估了格列本脲(一種BCS II類藥物)在生物相關介質中的溶解度。研究表明,當應用計算機模擬時,在膽鹽介質(含有牛磺膽酸鈉和卵磷脂)中增加的溶解度適合于建立體內-體外相關性(IVIVIVC)。在GastroPlusTM中建立了線性回歸。參比制劑的回歸系數為0.94。當使用生理性介質的溶出度值輸入GastroPlusTM時,預測血漿Cmax和AUC的誤差(%)分別為7%和14%。在水性介質(pH 6.5)中獲得的溶解度相對于血漿Cmax和AUC的預測誤差分別為38%和63%。后來,在生理相關介質(即FaSSIF)中開發了動態溶出方案,該方案再次顯示出建立IVIVC的預測能力。然后將此動態流通裝置溶出方案應用于孟魯司特鈉。同樣,生理性介質與臨床觀察數據的擬合最好。這些早期研究在不考慮其他胃腸道因素的情況下成功地建立了IVIVC。在Almukainzi等人的一項研究中,研究了胃動力對美洛昔康藥代動力學(PK)的影響。觀察到兩種制劑(常規制劑與快速溶出制劑)在嚙齒類動物模型中給藥時具有相似的PK結果。然而,當胃動力受損時,胃控制了藥物的釋放,因此,常規劑量的藥物吸收是不同的。但是快速溶解制劑的PK接近健康狀態下的結果。本研究表明,處方組成的差異在健康狀態下不影響,但在疾病狀態下可能導致顯著差異。這項研究表明,在疾病條件下,胃能夠對PK參數(如血漿Cmax和Tmax)產生負面影響,此外,人們普遍認為,對于許多藥物來說,胃排空會影響空腹與餐后的PK。然而,很少有人關注GI運動對Cmax和Tmax的影響,這取決于給藥時間和MMC階段。這可能是由于用于量化和描述藥物的PK行為的PK模型縮小了平均PK中單個的變異性。然而,如果同時監測腸蠕動和PK,則觀察到的血藥水平與腸蠕動之間的關系越來越顯著。另一個影響藥物吸收的因素是腸液的成分。胃腸道中的緩沖系統是以碳酸鹽為基礎的。在常規的藥品質量控制和開發中,磷酸鹽緩沖液起主要作用,而碳酸鹽緩沖液很少使用。選擇磷酸鹽而不是碳酸氫鹽似乎會影響腸溶制劑的體內性能。早期的報道顯示腸溶產品在體內失敗(1964年),直到今天的幾十年里,體內研究才證實了這一點。此外,有證據表明,磷酸鹽和碳酸鹽緩沖液與腸溶包衣的相互作用似乎不同。很明顯,重新評估已建立的體外測試方法對于獲得體內相關性能以避免產品失敗非常重要。除了緩沖液的性質外,下一個重要的區別是體外溶出方案中使用的緩沖液濃度與胃腸道中目前的緩沖液濃度以及藥物溶出對腸道吸收的影響。多年來,兩相溶出作為評估藥物制劑體內性能的替代品。基于藥物在有機層中的的滲透量,可以進行與所吸收相關的預測。然而,它對IVIVC的影響尚未得到充分認識。在最近的一項研究中,我們研究了布洛芬在藥典以及與GI濃度相當的磷酸鹽緩沖液的溶出行為。結果表明,布洛芬在藥典條件下在不到15分鐘的時間內完全溶解。然而,在低緩沖液濃度下,這一過程需要更長的時間,并且由于布洛芬的羧酸性質,介質的pH值發生了顯著變化。然而,如果進行兩相溶出試驗,pH值隨著時間的推移恢復到初始值。這再次證明了適應生理的體外測試對于獲得體內狀態是多么重要。只有這樣才能確保體外方法能夠預測體內性能。未來需要對這些方法轉化為QC方法進行更詳細的研究。最后一個方面涉及藥物在體內的性能與體外行為無關。右美沙芬就是一個罕見的例子。這種藥物在2小時內被吸收到80%以上,但需要大約15-20小時才能觀察到Cmax。經典IVIVC將吸收與溶出相關聯。然而,在這種具體情況下,IVIVC會產生誤導。藥物在腸道中快速溶解,并在15分鐘內完全溶解。如前所述,80%的藥物在2小時內被腸細胞吸收。藥物在進入腸細胞后會被溶酶體吸收。作為弱堿,它在細胞質的生理pH值下具有高度親脂性。由于藥物會從腸細胞的頂端遷移到基底外側,它可以穿過溶酶體的膜,進入ph值為微酸性的水環境。在這個細胞器中,弱堿變得更親水,因此會被溶酶體包圍。這就是為什么它在血液中出現的時間比它被吸收的時間要長。應該指出的是,對于這些特定的藥物化合物,溶出度試驗不能表征體內性能,因為藥物產品的溶出度不能與血藥水平直接相關。是生物系統及其特定環境和細胞室之間的藥物分配決定了藥物在中央室的濃度,而不是藥物的溶解。總之,胃腸道藥物吸收受到不同生理因素的高度影響。體外性能測試應考慮并包括適應生理環境的測試方法,以確定潛在的臨床相關的劑型因素。BCS分類系統包括酸、堿和中性分子,可以幫助識別這些不同口服藥物吸收的潛在障礙。為了滿足所有這些標準,一個潛在的體外裝置,可以模擬不同的胃腸道條件,如圖3所示。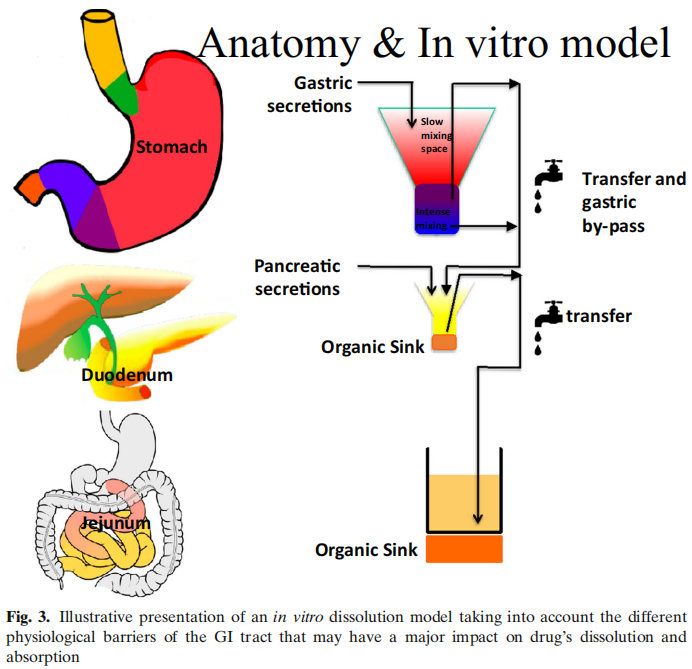
當兩種藥物不屬于同一BCS分類時,即當它們吸收的限制因素不同時,固定劑量藥物組合(FDC)產品的開發可能具有挑戰性。在介紹的第一部分中,在不同分類體系的框架內討論了探索組合產品中每種藥物的生物藥劑學特性的相關性。BCS標準框架,其中主要關注的是確定體外方法的非生物等效性(BE不等效)風險,這有助于處方的選擇。為了理解藥物的生物藥劑學的限制因素,在可開發性分類系統(DCS)需要去修飾它的滲透性與溶解性及BCS。DCS考慮了小腸中較高的可用液體體積(500 mL)和人體腸液中的溶解度及溶解性分類。500mL的體積是基于共同給藥時的送藥液體體積以及胃腸道中液體體積計算的。另一個相關的補充是區分溶解度受限和溶出度受限藥物,因為配方方法可能不同。可以根據藥物的物理化學特性選擇溶出試驗來探索體內不等效的風險。為此,Tsume等人提出了BCS的分類系統。BCS II類藥物分為中性(BCS IIc)、弱酸(BCSⅡa)和弱堿(BCSⅢb)。根據這些細分,用于預測體內行為的溶出試驗不同于I類和III類,其簡單的溶出儀(如USP II)就足夠了,而II類和IV類則應包括隔室和吸收池,以增加體內的可預測性。為了適應這種需要,文獻中提出了幾種溶出體系,并描述了幾種兩相或多相的可切換的溶出體系。
在第二部分,討論了制劑賦形劑的潛在影響,以及研究這些影響的實驗臨床前模型。在胃排空或腸道上,賦形劑可以影響膜通透性、代謝和胃腸道運動。在表I中,總結了一些具有有用參考文獻的實驗方法。
例如,十二烷基硫酸鈉(SLS)對非索非那定腸通透性的影響通過Doluisio的閉環灌注法進行了表征,并在大鼠體內BA研究中得到了進一步證實,而在大鼠胃排空試驗中,通過硫酸鋇評估了由于賦形劑引起的胃排空變化與生物等效性(BE)研究失敗的相關性。最后,使用BCS作為BE問題的風險評估工具的概念是在FDC開發的案例研究的幫助下提出的。纈沙坦/氫氯噻嗪仿制藥在BE測試中兩次失敗,其中一種藥物失敗,另一種藥物成功。使用胃腸道模擬器(GIS)進行的生物預測溶出試驗的應用成功地再現了體內結果,因為由于制劑中山梨醇和SLS的不同水平,胃室內崩解的差異和腸道中溶出速率的差異是體內失敗的顯著原因。總之,FDC中藥物的BCS和/或DCS分類是定義吸收影響因素和影響BA的相關生理變量的工具。對于含有不同BCS類別藥物的FDC,有必要結合體外溶出方法和臨床前模型來評估制劑性能。
FDC產品以固定劑量比將兩種或兩種以上活性藥物成分(API)組合成藥物劑型。FDC產品是根據組合規則批準的,該規則規定每個成分都應有助于產品的有效性,并且該組合在特定患者群體中也應是安全的。安全性和有效性數據可以是全部(新藥申請)或部分(505(b))原始數據,也可以基于以前的報告(簡稱新藥申請)。FDC產品比單一實體產品(SEP)的聯合給藥具有幾個優勢,如最大的患者依從性、提高的安全性和有效性、最大限度地減少濫用潛力以及降低患者成本。它們還為制造商提供了在產品生命周期中擴展知識產權和排他性的機會。另一方面,配制FDC產品帶來了一些與原料藥之間的不相容性以及與某些賦形劑的不相容相互作用相關的挑戰。一些藥物可能在另一種藥物(鹽酸胺碘酮-青蒿琥酯)存在下降解,另一些藥物可能具有藥物不相容性(辛伐他汀-替米沙坦),一些藥物可能表現出非常不同的粘彈性特性(二甲雙胍-格列本脲),以及其他可能在吸收(例如,腸道轉運蛋白)或吸收后(例如,代謝酶、腎轉運蛋白)水平上的相互作用。
世界衛生組織根據產品注冊的監管要求,將FDC產品分為四種不同的情況:
①新的FDC產品具有與現有FDC產品相同的活性成分和劑量。
②新的FDC產品與單一實體產品(SEP)的具有相同的活性成分和劑量。
③新的FDC產品將具有既定安全性和有效性數據的原料藥組合在一起,但尚未用于該特定適應癥。新FDC產品包括具有既定安全和有效性的原料藥的組合,但將用于不同的給藥方案。
④新FDC產品含有一種或多種新的化學實體(NCE)。
為了將參比制劑(RLD)的關鍵臨床數據與屬于上述①和②的FDC產品的安全性和有效性聯系起來,需要進行BE研究。雖然①的BE研究設計是標準方法,但在②中,將FDC產品的體內性能(例如,PK終點)與sep的聯合給藥進行比較。在這兩種情況下,成功的BE表明API之間不存在(或類似)PK相互作用。然而,FDC產品的BE研究具有挑戰性,因為(i)組合產品中PK受試者內在的變異性;(ii)不同濃度的非線性PK;(iii)藥物制劑的相互作用;和(iv)當作為組合產品服用時,食物對活性成分PK的不同影響。這些考慮使生物豁免成為制造商滿足BE要求的極具吸引力的機會。目前,WHO、FDA、歐洲藥品管理局(EMA)、國際協調會議(ICH)和加拿大衛生部允許對僅含高溶解度API的立即釋放(IR)FDC產品使用基于BCS的生物豁免。因此,含有BCS I類和/或III類活性成分的FDC產品可以申請生物豁免。
一般來說,溶解和組成要求與SEP的要求相同,但各司法管轄區之間存在一些差異。對于BCS I類API 的產品,FDA要求使用目前FDA批準的IR產品中存在的輔料,而EMA鼓勵使用與參比制劑相似量的相同輔料。2018年ICH關于基于BCS的生物豁免指南指出,關鍵賦形劑(如聚山梨醇酯80、山梨醇)必須在參考產品的±10%范圍內。另一方面,各司法管轄區對輔料可能對BCS III類藥物產生的影響達成共識,例如各機構要求輔料在種類上(Q1)與參比相同,在數量上(Q2)與參比非常相似。FDA和ICH指南包含賦形劑(按功能)相對于參比制劑的成分允許差異表。如果產品是藥物等效物,并且滿足溶解和成分要求,則對①中FDC產品實施基于BCS的生物豁免是簡單的。如果產品滿足溶解和組成要求,則對①中FDC產品實施基于BCS的生物豁免是容易實現的。此外,如果有適當的理由,美國食品藥品監督管理局可能會接受基于BCS的仿制藥生物豁免。另一方面,基于BCS的②中FDC產品生物豁免給制造商和監管機構帶來了一些挑戰。首先,不同的單一實體RLD產品可能在不同的地區注冊,這意味著制造商必須進行多次生物豁免研究才能在不同的司法管轄區獲得批準。與FDA不同,EMA沒有公布不同歐洲國家的RLD清單,這可能會進一步復雜化。其次,含有不相容原料藥的FDC需要結合分離技術(例如,雙層片、片中片等),以獲得穩定的產品。在這種情況下,可能很難解釋FDC產品和各自SEP之間的成分要求。第三,研究活性成分劑量差異較大(即劑量比>50%)的FDC產品的溶出方法在分析上可能具有挑戰性。當API在生理范圍內表現出不同的pH依賴性穩定性的情況下,這可能會更加復雜。此外,RLD SEP可能使用不同的溶出裝置(如籃或槳),因此制造商可能必須為一種FDC產品開發和驗證兩種溶出方法。第四,FDC產品中可能存在藥物-藥物或藥物-輔料PK相互作用(DFI)的因素,當SEP共同給藥時,這些相互作用可能不同或不存在。美國食品藥品監督管理局和歐洲藥品管理局都發布了關于在轉運蛋白水平上研究藥物相互作用(DDI)的指南。美國食品藥品監督管理局還發布了研究體外轉運體介導的DDI的方法學建議。雖然各機構要求申辦者研究腸外排轉運體介導的DDI(即p -糖蛋白、乳腺癌耐藥蛋白),但目前尚無關于研究腸攝取轉運體介導的潛在DDI的公開建議。這似乎令人驚訝,因為眾所周知,腸道表達的攝取轉運蛋白與大量結構不同的化學和治療類藥物有相互作用。此外,越來越多的證據表明,藥物輔料可以在體外和原位抑制腸道轉運蛋白的外排和攝取。包括聚乙二醇類表面活性劑,司盤和聚乙二醇。
雖然人們一致認為DDI或DFI可能與BCS I類藥物的臨床相關性較小,但也有人擔心這些相互作用可能會極大地影響低滲透性藥物的口服吸收。FDC產品還為開發一系列可用于通過劑量滴定優化治療的給藥方式提供了機會。中等規格和低規格可適用于基于劑量規格(DS)的生物豁免,前提是至少有一種規格(通常是最高的)在體內成功證明了與參比制劑的BE等效。基于劑量規格的生物豁免適用于不符合基于BCS的生物豁免條件的原料藥和IR以外的藥物形式(即改良釋放、延遲釋放)。各司法管轄區對基于DS的生物豁免的共同要求是治療劑量范圍內的線性PK,有機會在最高和最低規格之間采用括號法,并且制造過程相同。FDC的劑量范圍將取決于研究藥物的相加或協同作用。藥物之間的相互作用在藥物-藥物相互作用和PK-PD研究中進行評估。隨后,暴露反應模型可用于2B期劑量選擇。與基于BCS的生物豁免一樣,基于DS的生物豁免要求是SEP的擴展。表II和表III總結了基于DS的生物豁免的FDA和EMA的要求和溶出方法建議。表II和表III中的數據表明,在美國和歐洲市場尋求基于DS的生物豁免的制造商在滿足基于分離技術的FDC產品(如雙層片劑)的成分要求方面可能面臨挑戰,因為EMA將每一層視為一個單獨的實體,而FDA將雙層片劑視為單個單元。此外,在單一單位的情況下,原料藥之間劑量差異較大的FDC產品可能很難滿足美國食品藥品監督管理局和歐洲藥品管理局的比例要求。更具體地說,EMA規定,為了計算API/輔料的比例,必須將其他API視為輔料。然而,尚不清楚是否必須將其他API視為輔料。同樣,對于如何在雙層片中考慮其他API,FDA也沒有具體的建議。這些差異可能會阻礙FDC產品在美國和歐洲同時注冊申報。此外,雖然美國食品藥品監督管理局要求對不同規格中的最高劑量進行BE研究,但歐洲藥品管理局要求除最高規格外,還需要對最低規格進行研究。最后,不同司法管轄區之間存在不同的參比制劑,增加了申辦方在不同地區尋求批準時需要的不同的研究數量。
Amitava Mitra 博士(Sandoz,Inc.,A Novartis Division)討論了在實現含有兩種或兩種以上活性成分的FDC產品的BE過程中克服挑戰的關鍵點和策略。這些產品的活性成分可以通過不同的藥理學途徑發揮作用,并具有增加/協同作用、減少每種活性成分的劑量和提高患者依從性的優勢。帕金森氏癥藥物左旋多巴(Levodopa)的新型fdc是利用新技術和新機制改善舊藥物臨床療效的一個例子。
然而,將多種活性成分組合可能會使其單獨的生制藥和PK行為復雜化。由于藥物釋放的不同,控制或修飾釋放FDC產品的開發確實增加了額外的挑戰。釋放曲線的變化可能會改變API的生物藥劑學和藥代動力學。還討論了嚴格審查FDC中單個藥物的物理化學和生物藥劑學特性及其對PK的影響的重要性。全面了解單個藥物的PK特性以及FDC中的配方變量是同樣重要的需要考慮的因素。與FDC戰略相關的BA研究非常重要,值得鼓勵。然而,有太多變量的且不充分研究可能會進一步混淆已經很復雜的問題,應該避免。在設計BE研究時,應充分考慮來自所有化合物的所有物理化學、生物藥劑學和PK數據。可以考慮針對高變異藥物的BE研究設計,如收緊BE或交叉重復設計。利用從不同但協同的技術中獲得的知識,如體外溶解度/溶解性研究,吸收模型和IVIVC,體內臨床前動物模型,以及可用的體內臨床數據,對于給定組合的FDC策略的成功至關重要。討論了兩個案例研究,其中使用口服吸收建模、溶出度數據和臨床PK數據成功開發FDC產品。在第一個案例研究中,討論了三聯產品的開發,其中一種活性成分具有高度可變的Cmax,而另一種活性成份由于膽汁分泌和吸收緩慢而具有較長的Tmax。在這種情況下,口服吸收建模是理解配方變化對三種活性物質PK的影響以及最終對FDC產品開發的影響的關鍵點。在第二個案例研究中,討論了雙組份產品的開發,其中一種活性成分是弱堿,具有高的受試者體內CV和陡峭的pH溶解度。在這種情況下,來自幾項相關BA研究的數據以及對PK和生物藥劑學特性的深入了解有助于FDC的成功開發。
盡管其中一個目的是以固定的劑量比組合藥物,以簡化慢性病的治療并提高患者的依從性,但人們普遍認為,這一原理不能成為任何開發或配方設計背后的唯一目標。對世界各地司法管轄區FDC產品備案法規的概述表明,這方面的進展相當緩慢。總的來說,通過結合先前批準的單產品或從NCEs的共同配方開始開發FDC產品可以遵循有限的監管途徑。根據美國法規,FDC的監管基本原則在《聯邦法規》和指南中進行了描述,概述了FDC產品批準的要求。2013年引入的開發指南從監管角度反映了這些藥品的重要性。該指南描述,如果新FDC的劑量、擬定的治療適應癥、或者所需要的臨床數據沒有變化,則藥物療效可以依賴于BE測試。FDC產品可以遵循以下監管途徑之一:505 b(1)、505 b(2)或505 j,涵蓋從新藥開發到仿制藥開發的所有可能性。另一方面,EMA發布了關于FDC產品臨床開發的若干指南,指明了任何FDC開發的擬治療用途和適應癥。該指南描述了三種可能的情況,并對療效證明提出了具體要求:(1)如果對擬組合中包含的一種或多種藥物存在不良反應,則使用FDC產品作為附加治療。如果聯合用藥對潛在的臨床后果構成威脅,則可能需要進行藥物-藥物(DDI)或PK的相互作用研究,(2)當尋求減少藥片數量負擔時,用FDC產品替代。如果FDC產品以不同的時間間隔給藥,則需要進行BE測試,并應特別注意;如果FDC以前沒有用于任何特定的適應癥,開始FDC治療,則臨床和pk試驗以及DDI研究都應在批準前進行并提交。在拉丁美洲,自2010年以來,FDC產品的注冊只有一個具體指南,它描述了FDC產品的定義、申報的一般考慮以及取決于擬定劑量方案或擬聯合藥物的監管要求。在以下條件下可以獲得FDC批準:(1)FDC產品含有與同時使用的單一產品相同的活性物質、劑量和給藥方案;因此,安全性和有效性是眾所周知的;為了證明療效,BE研究可能就足夠了;(2)與“(1)”中的條件相同,但FDC產品將用于新劑量或新的治療適應癥,因此需要進行III期臨床試驗;(3)該組合含有一種或多種新的活性成分,需要進行I、II和III期臨床試驗才能獲得批準。一般來說,對于FDC產品的注冊沒有全球適用的指南,但針對特定的治療類別和世衛組織技術報告中描述的四種一般情況,旨在指導制藥公司在欠發達司法管轄區開發、批準和銷售FDC產品。盡管在大多數司法管轄區,仿制藥和復方藥提交途徑似乎已經足夠多,但是新的復方制劑仍然需要臨床前與臨床數據即使FDC藥物中單個的藥物是已知的或者是新發現的。然而,監管者仍然堅持認為,世界各地不同的司法管轄區應更加重視便利性/合規性,將其作為開發FDC產品的基本原理,要么包含授權/新藥實體,要么只考慮患者滿意度或降低/控制醫療成本。如果允許仿制藥開發,FDC產品的BE研究設計應考慮與單獨給藥相同的原則,尋找每種FDC活性成分及其各自的參比FDC或參比單品的PK的等效性。在一點上,這是十分重要的意識到PK相互作用可能會有更加關鍵的結果在FDC產品上相比于同時給予相同藥物的單一產品。總之,在比較獲得FDC產品批準的司法管轄區時,似乎有必要在FDC產品過于謹慎的注冊、有效性證明與FDC產品可能帶來的巨大公共衛生效益之間取得平衡。在FDC產品開發、注冊的現有技術指南、理解和應用方面實現全面的平衡仍然是一個未解的問題。
對于沒有FDC開發經驗的配方科學家,討論了兩個決策樹,以選擇最合適的配方開發策略。第一個決策樹與FDC產品的配方設計有關(圖4)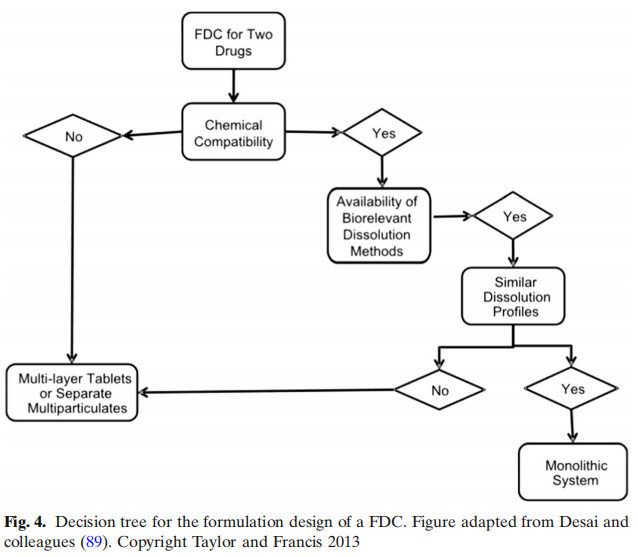
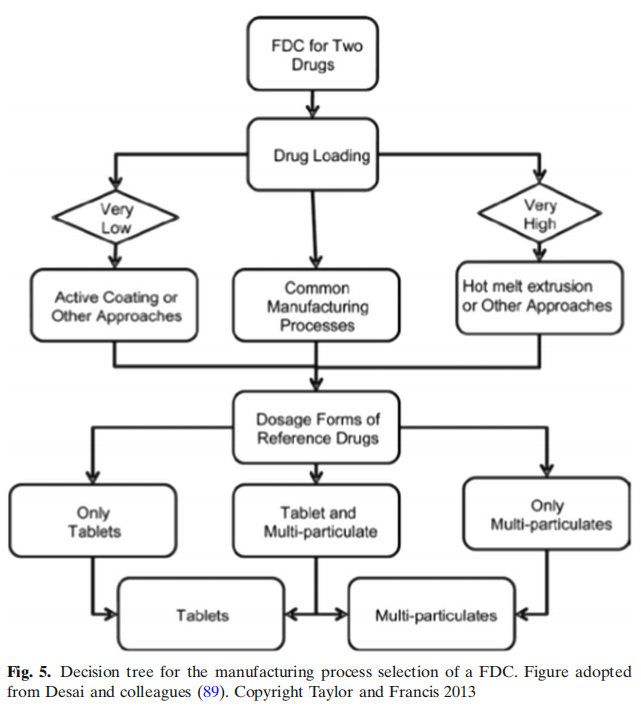
制劑中的藥物載藥量決定了生產工藝的選擇。如果載藥量大,可采用熱熔擠出或雙層法制備。對于低載藥量的制劑,提出了一種活性成分上藥包衣方法。制造工藝選擇的關鍵因素之一是制藥科學家先前的經驗。單片制劑系統,即兩種藥物合并在一個單劑量單位中,被認為是最簡單的制劑方法。然而,在一個案例研究中,第二種藥物氫氯噻嗪(HCTZ)被添加到現有的高血壓藥物配方中。結果表明,聚維酮(一種粘結劑)和波洛沙姆(一種潤濕劑)通過在有效水分中溶解HCTZ,在加速儲存條件下引發HCTZ降解。用淀粉1500代替聚維酮,解決了穩定性問題,并去除了泊洛沙姆,對BE研究沒有不利影響。對于雙層片劑處方方法,通常用于將兩種不相容的藥物分開或保持兩種藥物釋放曲線,很少有關鍵的處方因素被提出。這些因素包括選擇具有高脆性的賦形劑,如第一層中的乳糖;第二層中的可塑性材料,如微晶纖維素,以及兩層的重量比不超過1:6。還強調的是,第一層的壓力應該能夠在不犧牲表面粗糙度的情況下減小體積,而表面粗糙度對于第二層的粘附是必不可少的。提出了關于雙層片處方方法的兩個案例研究。在第一個案例研究中,通過添加1% w/w的二氧化硅,二甲雙胍緩釋制劑的可壓縮性得到改善。在第二個案例研究中,兩種不同等級的氣相二氧化硅在雙層片劑配方中的表現不同。Aerosil 200沒有引起層分離,但Aeroperl 300引起了分層。由于其更大的表面積,在任何濕度水平下,Aeroperl都能吸附相對大量的水分,但當濕度降低時,它不能保留水分。相比之下,Aerosil吸附的水分相對較少,但由于其大孔徑,它能保持水分。據推測,Aeroperl未保留的水分可與其他層賦形劑(如交聯聚維酮)相互作用。提出的第三種配方技術是包衣技術。包衣層也可用于維持兩種單獨的釋放曲線,并分隔兩種不相容的藥物。一個案例研究展示了酸堿敏感分子是如何穩定的,選擇可以降低API與輔料密切接觸包衣材料。例如,對于100mg片劑,將1mg藥物與99mg賦形劑一起放置,1mg的藥物可以與99mg的賦形劑反應;然而,如果將1mg的藥物與9mg的包衣材料一起放置,則可用于反應的量急劇減少。這也是生產壓縮敏感藥物片劑的有用技術。雖然活性涂層是有用的,但它沒有像其他技術那樣廣泛應用,因為它提出了兩大挑戰。第一個挑戰是如何檢測包衣終點,從而生產出具有擬定作用的片劑。如果包衣過程過早停止,片劑可能藥效不足。另一方面,如果涂層停止得晚,片劑可能會超級有效。第二個挑戰是含量均勻性(活性成分涂層均勻性)。含量均勻性可能受到各種工藝參數的影響,如包衣鍋生產批量、包衣時間、噴槍的數量和包衣質量。提出了模型參數與工藝參數相聯系的數學模型。結果表明,該模型能準確預測200 ~ 1450mg不同形狀片劑在450kg商業規模下的包衣均勻性。總之,決策樹對于探索FDC最合適配方和制造過程非常有用。FDC的每種配方方法都有其獨特的挑戰,但正如各種案例研究所示,有可能克服這些挑戰,利用各種過程分析技術(PAT)開發堅固的配方和商業上可行的制造工藝。
組合藥物在《聯邦法規法典》21 CFR 3.2 (e)中定義為藥物-藥物聯合產品的類別。這些產品可以是兩種已批準藥物或者兩種已批準以上藥物或者是在研藥物與已批準藥物或者兩種在研藥物或者兩種以上在研藥物組合在一起共同開發。最終產品可以是FDCs、共包裝產品或分開的單個的藥品再一起使用。開發這些產品的原因之一是針對同一疾病的藥物(例如抗病毒和咳嗽/感冒藥產品)的加性/協同效應。開發這些產品的原因之一是針對同一疾病的藥物(例如抗病毒與咳嗽/感冒藥物產品)的相加/協同作用。有時,當兩種藥物具有互補的作用機制時,它們會被作為FDC產品開發用于同一疾病。例如,將β內酰胺與β內酰胺酶抑制劑相結合,可以選擇性殺死對β內酰胺具有耐藥性的細菌。有一些FDC的例子,某成分減少其他成分(例如萘普生/埃索美拉唑緩釋片)的不良事件。大多數FDC是口服的,但也有吸入性的例子(例如,用于慢性阻塞性肺病(COPD)的噻托溴銨/奧達特羅)與眼科產品(例如用于降低眼壓的奈舒地爾/拉坦前列素)。本報告的目的是概述FDC開發中的臨床藥理學考慮。FDC的開發給藥物開發人員帶來了有趣的挑戰。如果正在開發兩種或兩種以上的新分子實體(NEMs)作為FDC,則通常需要對藥物給藥劑量進行研究,以確定每種藥物的合適的給藥劑量。藥物管理方面的挑戰,如食物對FDC的影響,通常需要加以解決。當FDCs中提出的各種藥物在進食和禁食條件下的給藥要求不同,或者藥物給藥頻率不同時,這種情況可能會變得更加復雜。這些情況通常需要更仔細地研究FDC的配方和進行額外BA研究。考慮到配方的不靈活性,特定人群中FDCs的劑量調整可能存在問題。作為FDC開發計的典型研究是關乎BA研究。FDC BA研究的目的是將FDC中每種活性藥物成分或治療部分的吸收速率和吸收程度與作為單獨的單成分制劑同時給藥的的吸收速率、吸收程度進行比較21 CFR 320.25(g)。通常,建議在復方產品最高規格并且與相匹配的單個藥物的制劑下對比研究復方制劑與單活性成分藥物的二周期、單劑量空腹研究。替代性研究設計,如比較聯合用藥產品與單一成分藥物產品的單獨給藥的治療研究設計,也可能是合適的。單劑量、食物影響這樣在FDC產品上的研究用來評估食物對FDC的影響。本文介紹了與支持FDC批準的研究相關的BA研究案例,以及基于析因設計研究的FDC與單獨給藥的例子。FDA指南題為“聯合開發兩種或兩種以上的新藥”,列出了可能適合進行因子設計研究以確定FDC中單個成分的作用的情況。
FDC產品的市場準入具有挑戰性,一方面是要實現單個藥品的協同給藥,另一方面也是由于配方方面的挑戰(原料藥的相容性、劑量)。然而,我們不應忽視胃腸道生理學對口服藥物行為的影響,這可能導致受試者間差異,從而可能導致BE研究的失敗。因此,在監管當局的指導方針的支持下,確定制劑開發戰略與臨床評價之間的關系是很重要的。此外,體外溶出測試的貢獻可以在某種意義上幫助監管機構對FDC產品批準的決策,因為這些模型可以模擬在胃腸道吸收過程中起關鍵作用的潛在的GI變量。從學術角度來看,當制藥公司分享他們的非BE配方(即臨床失敗)時,可以對這些臨床相關的溶出模型進行優化和驗證。當他們這樣做時,潛在的問題可能被解開,配方科學家在制定FDC產品時應考慮到這些問題。






